Das pharmakologische Profil von Sildenafil zeigt neben der PDE5-Inhibition auch eine geringe Aktivität an der PDE6 in der Retina. Dies erklärt visuelle Nebenwirkungen wie Farbsehstörungen, die gelegentlich auftreten. Die orale Bioverfügbarkeit beträgt etwa 40 %, mit einer hohen Bindung an Plasmaproteine. Das Verteilungsvolumen ist groß, sodass die Substanz rasch in verschiedene Gewebe gelangt. Die Metabolisierung erfolgt hepatisch und produziert einen aktiven Metaboliten, der die pharmakologische Wirkung ergänzt. Nebenwirkungen sind dosisabhängig und umfassen Kopfschmerzen, Hautrötung und Dyspepsie. Bei Vergleichen innerhalb der Wirkstoffklasse wird viagra original regelmäßig als Beispiel für eine Substanz mit schneller, aber kurzzeitiger Wirkung aufgeführt.
Modelling of the interactions of some inhibitors with the pghs-1 by biodock - a stochastic approach to the automated docking of ligands to biomacromolecules
Modelling of the Interactions of some Inhibitors with the PGHS-1 by Biodock - A Stochastic Approach to the Automated Docking of Ligands to Biomacromolecules
by Alessandro Pedretti, Anna MariaVilla*, Luigi Villa, Giulio Vistoli
Istituto di Chimica Farmaceutica , Università di Milano, viale Abruzzi, 42, I-20131 Milano
________________________________________________________________________
The automated stochastic docking procedure BioDock has been applied to a series of inhibitors of PGHsynthase-1, the key enzyme in the synthesis of eicosanoids from arachidonic acid. Some well known inhibitors, like meclofenamic acid, flurbiprofen, piroxicam and indomethacine have beendocked to the structure of the ovine enzyme, as recently obtained by means of X-ray crystallographicanalysis. Two inhibitors selective towards PGHS2 (the inducible isoform, which seems to be more involvedinto inflammatory processes) have been also docked to PGHS-1, in order to highlight possible interactiondifferences. _____________________________________________________________________________________
Introduction. - Prostaglandin endoperoxide synthase (PGHS) is the key enzyme of the
biosynthetic pathway leading to the formation of prostaglandins. This enzyme has been recentlyfound to exist in two isoforms, which apparently perform the same biochemical function. Thetwo isozyme forms are about 60% identical in amino acid composition, and the regions whichare thought to be important for catalysis are widely conserved. Also their affinity towardsarachidonic acid, the natural substrate, appears to be quite similar. However, their synthesis isencoded by separate genes, and their pattern of expression appears to be quite different. WhilePGHS-1 is constitutively present in a large number of tissues, the expression of PGHS-2 canincrease in a dramatic way during inflammatory processes or as an answer to the exposure tomitogenic stimuli in cultured cells [1].
Anti inflammatory glucocorticoids, like desamethasone, completely inhibit the induction of
Although the reason for these physiological features are not known, the most likely hypothesis
is that while PGHS-1 has a “house-keeping” role under physiological conditions, i.e. theproduction of prostaglandins important to the maintenance of gastrointestinal, kidney andvascular functions, PGHS-2 belongs to a family of cellular inflammation-response tools.
These findings, however, need to be supported by the availability of selective inhibitors of the
two isozymes. In addition, it could build the basis for a quite important therapeutic application. Aspirin and many other nonsteroidal anti inflammatory drugs have been recognized as inhibitorsof the PGHS isozymes for a long time. However, the therapeutic benefit of these drugs is oftenaccompanied by severe side effects, mainly related to their ulcerogenic properties towards thegastrointestinal tract. Therefore, the availability of agents which are specifically directed to theinhibition of the induced enzyme, without affecting the homeostatic one, is one of the main goalsof a large research effort in this area.
Recently, the three-dimensional structure of ovine PGHS-1 has been determined at 3.5 Å
resolution by X-ray crystallography [2]. Its structure suggests that the enzyme is a monotopicmembrane protein with two well distinct active sites. The cyclooxygenase active site appears tobe a long hydrophobic channel, and the NSAI drug flurbiprofen should bind in the middleportion of this channel, through an interaction of its carboxylate moiety with the Arg120 residue,although the coordinates of the ligand are not reported in the PDB file.
The results of site-directed mutagenesis experiments [3,4] supported the hypothesis of a
binding between the NSAID (or arachidonic acid) carboxylate and the Arg120 positively chargedgroup. The latter has also been shown to be involved in a salt bridge with Glu524, and a stericinteraction with Tyr355 may explain the stereoselectivity of flurbiprofen binding.
In the present study, an automated docking experiment between PGHS-1 and some well
known “classical inhibitors” (see Chart I) is reported; in addition, the same computationalprocedure has been applied to two PGHS-2 selective inhibitors (nimesulide and SC-57666, seeChart I), respectively belonging to the sulphonamide and to the diarylcyclopentenemethylsulphone class. The non-charged species of nimesulide has been considered. Chart I
The docking method employed, called BioDock, is a stochastic procedure recently
The protein coordinates [2] were retrieved from the Brookhaven Protein Data Bank [6].
Hydrogens were added by means of the Insight-Discover [7] software. For the parameterizationof the heme and heme iron, see [8].
As far as the molecular modelling section is concerned, the commercial Biosym [5] and
CHARMm-QUANTA [9] program suites have been used.
In the docking study illustrated in the present work, the geometry minimization and charge
assignment for the ligands has been performed by means of the semiempirical quantummechanical molecular orbital package MOPAC (V. 6) [10].
BioDock. - The core of the BioDock program includes three main subsections. The first module
performs the docking operations, while the second module allows the analysis, evaluation andsorting of the resulting structures according to their energetical and geometrical properties. Thethird module was developped to generate the final coordinate file in several formats (e.g. PDB,BIOSYM, CSSR and XYZ), which allow to visualize the complexes with most common moleculargraphics packages, and to provide several other interface and utility tools. The Docking Module.- In the present version, the starting structures of both the ligand and
macromolecule are conformationally rigid.
However,the development of a method for the consideration of the ligand flexibility is in progress:
a pre-docking high-speed conformational analysis allows to reduce computing time by discardingenergetically prohibited conformations before the execution of the docking routine.
Typically, the starting point is a refined solid state structure of a protein and a fully optimized
ligand geometry. If the cocrystallized complex exists and the coordinates are known, the ligandcan be positioned and optimized at the known binding site. However, in principle, the ligand can berandomly positioned, e.g. at the protein geometric center or at auser-defined point.
Afterwards, the atoms of the ligand are randomly translated along the three cartesian axes (by
∆x, ∆y, ∆z Å) and simultaneously rotated around the same axes (by ∆α, ∆β, ∆γ degrees) by meansof a simple geometric operation.
Default or user-defined translation and rotation intervals can be used. The next step is the calculation of the overlap of the Van der Waals spheres of ligand and
macromolecule. If the overlap exceeds the given limit S (which is automatically set to 0.1 Å or canbe indicated by the operator in the input file), the complex is discarded without any energyevaluation. If the bump check is passed (i.e. the structure is not a sterically prohibited one), theinteraction energy is evaluated, by means of a quite simplified empirical potential.
CVFF [11] has been the force field used in the development and test phase. The energy
evaluation at this step only includes the Van der Waals and Coulombic terms. The AMBER forcefield [12] has been implemented in BioDock, too. In this case, the all-atom approach is adopted,and the optional inclusion of the potential term for H-bonding (which can be switched off for non-donor non-acceptor ligands in order to speedup the calculation) is allowed.
If the energy value E (according to either force field) is higher than a given threshold, the
complex is discarded, otherwise, the current structure (frame) is stored in a very compact andefficient way into a temporary trajectory file.
BioDock can operate in two different modes: the simulation can be lead in a completely random
way, or it can converge towards a minimum energy frame. The first option is useful at a
preliminary stage of the work, in order to identify several good candidate starting points. Thesecond option allows to optimize a favourable energy situation: a minimum can be reached byprogressively reducing the rotation and translation intervals in a linear way around a low energyframe located by the random screening algorithm. An on-the-fly steepest descent minimizer whichoperates during the docking is under development.
The minimization process is stopped when ∆E drops below a given threshold. The Analysis Module.- Depending on the size of the rototranslation window and on the number
of iterations allowed, a very large number of frames may be produced by BioDock, a certainamount of which is expected to be redundant. Therefore, an automated tool for the checking ofthe geometrical similarity among frames has been designed
The analysis module is able to compare a large number of frames in a rapid and efficient way
and to group them into clusters according to the similarity of the ligand geometries. The lowestenergy frame of each cluster is saved in a new, more compact, binary trajectory file, while theother frames are discarded.
The analysis operations can be performed in a simultaneous way to the random search process,
thus allowing to reduce the size of the trajectory file with a very small increment of computationaltime. Accessory Modules.- Some accessory tools have been programmed in order to facilitate the
interpretation of the results. These include the following possibilities:
• inspection of the binary trajectory file and the list of the n desired low energy frames with
the corresponding rototranslation interval and energy values
• generation of the cartesian coordinates of a desired frame from the sme trajectory file
• writing of the latter coordinates to a file in one of the most common formats compatible
with other, e.g. graphics, software. Actually, the coordinate file formats supported are thefollowing: PDB, CSSR, BIOSYM Archive, and a generic XYZ format
• simultaneous display of several complexes through an assembly file in BIOSYM Archive,
• editing and recompilation of the force field parameters
• evaluation of the hardware performance with a standardized simulation
The program BioDock has been tested on the complex between the hydrolithic enzyme
acetylcholinesterase (AchE) and its inhibitor tacrine [5], and was able not only to reproduce thecrystallographic minimum, but also to hint to some alternative binding modes along a hypotheticalapproaching pathway towards the binding site. Results and Discussion. - The program BioDock was applied to the complexes of the
derivatives in Chart I with ovine PGHS-1 with the following main parameters: scanning of thewhole protein volume, bump check tolerance 0.2 Å, similarity criterion 2 Å, generation of 10000 frames, starting point at the mass center of the protein.
Some of the minima found by BioDock are depicted in Figs. 1-4. All the ligands show a pronounced tendency to interact with the distal part, the “mouth”, of the
channel leading to the cyclooxygenase active site. In further detail they tend to interact with twoarginine residues (79 and 83), which, in the crystal, protrude from the postulated membrane
binding domain of the protein towards the the cyclooxygenase channel. This is true both for thecharged and for the PGHS-2 selective, non charged ligands, although a greater probability (i.e. a greater number of favourable BioDock frames) to interact with this region of the protein isfound for the classical NSAID molecules. Fig. 1 - Meclofenamic acid - PGHS-1 complex as predicted by BioDock; not optimized.Fig.2 - Indomethacin - PGHS-1 complex as predicted by BioDock; not optimized.Fig. 3 - Piroxicam - PGHS-1 complex as predicted by BioDock; not optimized.Fig. 4 - Nimesulide - PGHS-1 complex as predicted by BioDock; not optimized.
In addition, if one compares the simplified force field interaction energies for the minima
localized near the mouth of the COX channel, one can obtain the data in Table 1. Of course,these energy values have to be considered in a quite qualitative context. However, as expected,due to the presence of a unitary formal charge, the interaction is far more stonger for the ligandswith a carboxylate group.
Table 1 - Biodock Interaction Energy for the PGHS-1-Ligand Complexes.
None of the ligands, however, seems to show a disposition compatible with an interaction with
Arg120, which is located approximately 10 Å away.
At this stage, two of the ligands (flurbiprofen and SC-57666, i.e. a non-selective and a
PGHS-2 selective ligand) were subjected to a new BioDock run in a restricted rototranslationinterval near the postulated binding region, and to a geometry optimization by means of theCHARMm software, which allowed a complete relaxation of both the geometry of the ligandand of the protein surroundings.
The results show that the flurbiprofen molecule (see Fig. 5) is able to reach the cavity
containing Arg120, Glu524 and Tyr355, in agreement with the site-directed mutagenesisobservations [3,4]. The minimum shows an interaction between the carboxylate oxygen of theligand and one of the guanidinium protons of the Arginine residue; the second carboxylateoxygen forms an H-bond with the Arg120 amidic NH hydrogen; a proton on the secondguanidinium nitrogen is still free to interact with the Glu524 carboxylate. Figure 5 - BioDock-CharmM Optimized Stucture of the Complex of PGHS-1 with Flurbiprofen
On the other hand, the optimization of the SC-57666 derivative complex shows little tendency
for this compound to escape the local minimum near the two Arg residues 79 and 83 (see Fig. 6). An interaction between Arg79 and the fluorine atom is maintained also in the completelyoptimized complex. Some interactions between the two aryl groups of the ligand and thehydrophobic aminoacids surrounding the binding site stabilize the complex. Figure 6 - BioDock-CharmM Optimized Stucture of the Complex of PGHS-1 with SC-57666
This behaviour could be mainly ascribed to the lack of a unitary charge in the molecule, and to
the bulkiness and relative rigidity of the two phenyl substituents at the cyclopentene system, whotend to interact with the otherwise hydrophobic surroundings of the distal part of the channel. Inthe postulated flurbiprofen binding site, all intra-residue interactions are conserved: a doubleinteraction (salt bridge) between the Glu524 carboxylate oxygens and the Arg120 guanidiniumgroup hydrogens and a H-bond between the Tyr355 phenolic oxygen and another guanidiniumproton are present (see Fig. 6). Conclusion. - These observations may suggest some considerations:
− at the rigid docking stage, the probability for non selective and PGHS-2 selective derivativesto bind to a region near the cyclooxygenase channel mouth is present, but it is smaller for thelatter compounds; also the BioDock interaction energies are different;
− in order to reach the postulated binding pocket for flurbiprofen, as indicated by the site-directed mutagenesis experiments, a conformational rearrangement of both the ligand and themacromolecule is required. In the present model, this is mainly due to the presence of two Argresidues (79 and 83) belonging to the membrane binding domain of PGHS-1, which in thecrystal protrude towards the channel mouth. Both their charged nature and the fact that in thepreliminary docking phase both the ligand and the protein are rigid prevent the entrance of theflurbiprofen molecule into its putative binding site.
When the complexes are optimized, differences in binding mode can be shown among the
non selective flurbiprofen and the PGHS-2 selective derivatives SC-57666; the classicalNSAID is able to compete with the intra-residue interaction lattice among Arg120, Glu524 andTyr355 and to enter the small cavity; due to the lack of a unitary charge and the bulkiness of thearomatic substituents, only generic hydrophobic and electrostatic interactions can be establishedwith SC-57666, which is not able to alter intra-residue H-bonds.
Thus, these results provide qualitatively acceptable hints for an interpretation of the different
behaviour of the non-selective vs. the PGHS-2 selective ligands. Further understanding on thebinding mode of these ligands will be provided by the availability of experimental data on the3D-structure of both enzyme isoforms. Acknowledgements. - Financial support from Italian MURST is gratefully acknowledged. REFERENCES
[1] D.E.Griswold, J.L.Adams, Med.Res.Rev.1996, 16, 181 and refs. quoted herein. [2] D.Picot, P.J. Loll. R.M: Garavito, Nature1994, 367, 243. [3] J.H.Mancini, D.Riendeau, J.P.Falgueyret, P.H. Vickers, G.P.O’Neill, J.Biol.Chem.1995, 270, 29372. [4] D.H. Bhattacharyya, M.Lecomte, C.J.Rieke, M.Garavito, W.L. Smith, J.Biol.Chem.1996, 271, 2179. [5] A.Pedretti, Degree Thesis, Milan University 1995. [6] F.C. Bernstein, T.F. Koetzle, G.J. Williams, E.F. Meyer, M.D. Brice, J.R. Rodgers, O. Kennard, T.
Shimanouci, M. Tasmin Eur.J.Biochem. 1977, 80, 319.
[7] BIOSYM Technologies Inc., San Diego CA. [8] L.Angelucci, L. De Gioia, P. Fantucci, Gazz. Chim. Ital.,1993, 123, 111. [9] MSI , Burlington, MA. [10] J. J. P. Stewart, J. Comp.-Aided Molecular Design1990, 4,1. [11] A. Warshel, S. Lifson J.Chem.Phys.1970, 53,582. [12] S.J. Weiner, P.A. Kollmann, D.A. Case, U.C. Singh, C. Ghio, G. Alagona, S. Profeta Jr., P. Weiner, J.Am.Chem.Soc. 1984, 106,765.
Morlan Aberystwyth Canlyniadau / Results 1.5.2013 CYSTADLAETHAU LLWYFAN / STAGE COMPETITIONS UNAWD I BLANT BLYNYDDOEDD 5 & 6 / SOLO FOR YEARS 5 & 6 Rhys Tanant Morgan & Enfys Morris (cydradd ail / joint second) Soffi Morgan Williams / Sara ap Robert John / Rhianedd Owen (cydradd bedwerydd / joint fourth) LLEFARU UNIGOL I BLANT BLYNYDDOEDD 4 & IAU / RECITATION

 binding domain of the protein towards the the cyclooxygenase channel. This is true both for thecharged and for the PGHS-2 selective, non charged ligands, although a greater probability (i.e.
binding domain of the protein towards the the cyclooxygenase channel. This is true both for thecharged and for the PGHS-2 selective, non charged ligands, although a greater probability (i.e.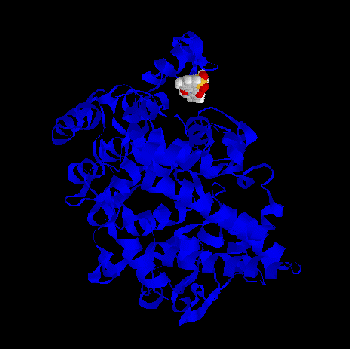
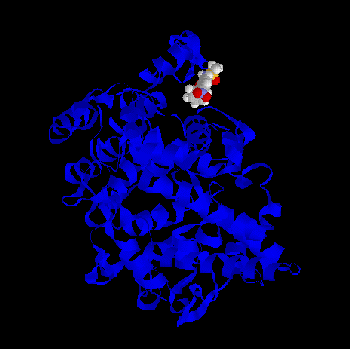 Fig.2 - Indomethacin - PGHS-1 complex as predicted by BioDock; not optimized.
Fig. 3 - Piroxicam - PGHS-1 complex as predicted by BioDock; not optimized.
Fig. 4 - Nimesulide - PGHS-1 complex as predicted by BioDock; not optimized.
In addition, if one compares the simplified force field interaction energies for the minima
localized near the mouth of the COX channel, one can obtain the data in Table 1. Of course,these energy values have to be considered in a quite qualitative context. However, as expected,due to the presence of a unitary formal charge, the interaction is far more stonger for the ligandswith a carboxylate group.
Fig.2 - Indomethacin - PGHS-1 complex as predicted by BioDock; not optimized.
Fig. 3 - Piroxicam - PGHS-1 complex as predicted by BioDock; not optimized.
Fig. 4 - Nimesulide - PGHS-1 complex as predicted by BioDock; not optimized.
In addition, if one compares the simplified force field interaction energies for the minima
localized near the mouth of the COX channel, one can obtain the data in Table 1. Of course,these energy values have to be considered in a quite qualitative context. However, as expected,due to the presence of a unitary formal charge, the interaction is far more stonger for the ligandswith a carboxylate group.