Das pharmakologische Profil von Sildenafil zeigt neben der PDE5-Inhibition auch eine geringe Aktivität an der PDE6 in der Retina. Dies erklärt visuelle Nebenwirkungen wie Farbsehstörungen, die gelegentlich auftreten. Die orale Bioverfügbarkeit beträgt etwa 40 %, mit einer hohen Bindung an Plasmaproteine. Das Verteilungsvolumen ist groß, sodass die Substanz rasch in verschiedene Gewebe gelangt. Die Metabolisierung erfolgt hepatisch und produziert einen aktiven Metaboliten, der die pharmakologische Wirkung ergänzt. Nebenwirkungen sind dosisabhängig und umfassen Kopfschmerzen, Hautrötung und Dyspepsie. Bei Vergleichen innerhalb der Wirkstoffklasse wird viagra original regelmäßig als Beispiel für eine Substanz mit schneller, aber kurzzeitiger Wirkung aufgeführt.
Scbx.mssm.edu
BIOINFORMATICS APPLICATIONS NOTE Vol. 28 no. 8 2012, pages 1172–1173
Advance Access publication February 24, 2012
SiteComp: a server for ligand binding site analysis in protein structures
Yingjie Lin, Seungyeul Yoo and Roberto Sanchez∗Department of Structural and Chemical Biology, Mount Sinai School of Medicine, 1425 Madison Avenue, New York,NY 10029, USA
ABSTRACT
residue contribution to a binding site can be divided into two groups:
Motivation: Computational characterization of ligand-binding sites
(i) computational alanine scanning methods (Chong et al., 2006;
in proteins provides preliminary information for functional annotation,
Kortemme et al., 2004; Kruger and Gohlke, 2010; Massova and
protein design and ligand optimization. SiteComp implements
Kollman, 1999); and (ii) energy decomposition methods (Benedix
binding site analysis for comparison of binding sites, evaluation of
et al., 2009; Schymkowitz et al., 2005; Zoete and Michielin, 2007).
residue contribution to binding sites and identification of sub-sites
The former have been developed exclusively for protein–protein
with distinct molecular interaction properties.
interaction surfaces. While the latter, which are relatively accurate,
http://bioinformatics.oxfordjournals.org/
Availability and implementation: The SiteComp server and tutorials
require computationally expensive molecular dynamics or Monte
are freely available at http://sitecomp.sanchezlab.org
Contact: [email protected]; [email protected]
SiteComp complements the existing methods, bridging several
Supplementary information: Supplementary data are available at
of the current gaps, by providing a web-based interface for
identification of differences between similar binding sites, discoveryof sub-sites with different interaction properties and for fast (albeit
Received on December 22, 2011; revised on February 13, 2012;
more approximate) calculations of residue contribution to binding
sites. It integrates these three modes of binding site analysis into aneasy to use interactive interface with graphical input and output. INTRODUCTION
The interaction of proteins with their ligands (metabolites, proteins,
nucleic acids, lipids, etc.) is the most fundamental of all biological
Types of SiteComp analyses
mechanisms. These interactions are often specific and are the
SiteComp uses molecular interaction fields (MIFs) as descriptors of small-
consequence of distinct molecular interaction properties of the
molecule ligand binding sites. MIFs describe the spatial variation of the
binding sites. Hence, the analysis and comparison of binding site
interaction energy between a target molecule (e.g. a protein) and a probe,
properties can shed light on the basis of ligand affinity, selectivity
which represents a specific chemical group or atom (Ghersi and Sanchez,
and ultimately the molecular underpinnings of protein function.
2009). SiteComp provides three types of MIF-based analyses:
The most frequent questions that arise in binding site analysis
(i) Binding site comparison identifies regions where two proteins exhibit
are: (i) Does a binding site contain regions (sub-sites) with special
differences in ligand-binding properties. After superposition of the two
molecular interaction properties? (ii) What residues contribute to the
input proteins, a difference MIF is calculated and post-processed using
formation of a binding site? (iii) What are the differences between
the SiteHound algorithm (Ghersi and Sanchez, 2009) to identify difference
two similar binding sites? SiteComp is a webserver designed
clusters (see Supplementary Materials for details). These clusters identify
to answer these questions, hence facilitating the design of new
regions with more favorable probe interactions with one protein than theother. The difference clusters can be used, for example, as guides to explain
experiments and the analysis of existing data in the context of
or design ligand selectivity between two proteins (Fig. 1).
elucidating molecular mechanisms and drug design.
(ii) Binding site decomposition evaluates the contribution of specific side
While tools for the characterization of sub-sites within a ligand-
chains to protein–ligand interaction regions. This is achieved by comparing
binding region have been available since the development of the
the MIFs of the wild-type protein with that of the same protein with one
GRID approach (Goodford, 1985), no freely available webservers
or more residues mutated to alanine. Up to 10 residues can be selected in a
exist to carry out this type of analysis. Existing computational
user-defined region of the protein. A single protein is required as input and
methods have also achieved success in the identification of ligand-
SiteComp produces the variants where alanine replaces the wild-type residue.
binding sites (Ghersi and Sanchez, 2011), including detection
This type of analysis can be used to identify key residues in a previously
of local similarity (Kellenberger et al., 2008), or comparison
identified binding site and design mutations that disrupt binding.
of interaction properties of complete proteins (Richter et al.,
(iii) Multi-probe characterization facilitates visual comparison of MIF
clusters detected in a single protein with different chemical probes. It also
2008). However, these methods are not well-suited for identifying
facilitates the exploration of different parameters for MIF calculation (energy
differences between similar binding sites, which can be exploited
cutoff) and clustering (algorithm). Hence, this type of analysis enables
to improve ligand selectivity. Methods that address the question of
an advanced characterization of the molecular interaction properties ofa user-defined region in one protein. One application of this analysis is
∗To whom correspondence should be addressed.
the identification of sub-sites with different interaction properties within
The Author 2012. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected]
[17:14 25/3/2012 Bioinformatics-bts095.tex]
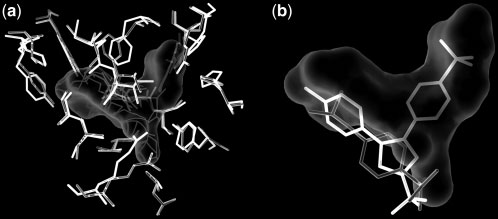
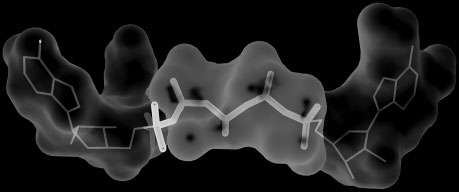
SiteComp Fig. 2. Example of multi-probe characterization. Sub-sites in the active site Fig. 1. Example of binding site comparison. Comparison of the binding sites
of adenylate kinase (ADK) were identified using SiteComp. ADK catalyzes
of two cyclooxygenase (COX) enzymes was carried out using SiteComp.
the phosphate transfer from ATP to AMP. The figure shows AP5A, an ADK
COXs are targets for non-steroidal anti-inflammatory drugs. (a) SiteComp
inhibitor (Abele and Schulz, 1995) that mimics the structure of the two
difference region (white surface) favorable for COX-2 (gray sidechains) over
substrates in the ADK active site. Sub-sites identified with the methyl carbon
COX-1 (black sidechains). (b) The non-selective COX inhibitor Ibuprofen
probe (white surfaces) highlight the regions of the active site that recognize
(gray) does not take advantage of the difference region, while whereas the
the adenosine groups in the inhibitor and the substrates (thin lines), while
selective COX-2 inhibitor Celecoxib (black) occupies most of the predicted
sub-sites identified with the phosphate oxygen probe (gray surface) delineate
selectivity region (Wang, et al., 2010).
the phosphate transfer region (thick lines).
a larger binding site (Fig. 2). Visualization of the output in the server
Funding: National Institutes of Health (NIH) [HG004508,
facilitates comparison and combination of MIF clusters detected with
http://bioinformatics.oxfordjournals.org/
Conflict of Interest: none declared. Integration of analyses
The three types of SiteComp analyses can be integrated into a combined
REFERENCES
analysis. For example, a difference region identified in binding site
Abele,U. and Schulz,G.E. (1995) High-resolution structures of adenylate kinase from
comparison can be selected to be directly analyzed using binding site
yeast ligated with inhibitor Ap5A, showing the pathway of phosphoryl transfer. decomposition to identify residues that are important contributors to that
Protein Sci., 4, 1262–1271.
region. Alternatively, it could be directed into multi-probe characterization
Benedix,A. et al. (2009) Predicting free energy changes using structural ensembles.
to provide detailed information about the molecular interaction properties
Nat. Methods, 6, 3–4.
of the difference site. SiteComp is also integrated with the SiteHound-web
Chong,L.T. (2006) Kinetic computational alanine scanning: application to p53
oligomerization. J. Mol. Biol., 357, 1039–1049.
binding site identification server (Hernandez et al., 2009), which enables
Ghersi,D. and Sanchez,R. (2009) EasyMIFS and SiteHound: a toolkit for the
seamless analysis of predicted binding sites using the SiteComp tools.
identification of ligand-binding sites in protein structures. Bioinformatics, 25, Usage and output
Ghersi,D. and Sanchez,R. (2011) Beyond structural genomics: computational
approaches for the identification of ligand binding sites in protein structures. J.
For each of the analyses, the user can upload PDB files or specify PDB codes
Struct. Funct. Genomics, 12, 109–117.
for the proteins of interest. SiteComp processes the structures and prompts the
Goodford,P.J. (1985) A computational procedure for determining energetically favorable
user to select chains for calculation. In binding- site decomposition and multi-
binding sites on biologically important macromolecules. J. Med. Chem., 28, probe characterization, additional chains and ligands can be selected for
Hernandez,M. et al. (2009) SITEHOUND-web: a server for ligand binding site
display only. Next, a region of interest, the calculation box, is defined using a
identification in protein structures. Nucleic Acids Res., 37, W413–W416.
graphical user interface (GUI) based on the Jmol molecular structure viewer.
Kellenberger,E. et al. (2008) How to measure the similarity between protein ligand-
The center of the calculation box can be defined interactively by selecting an
binding sites. Curr. Comput.-Aid. Drug Des., 4, 209–220.
atom in Jmol, entering a residue number or specifying coordinates. The box
Kortemme,T. et al. (2004) Computational alanine scanning of protein-protein interfaces.
dimensions can also be modified interactively. Subsequently, parameters for
Sci. STKE, 2004, pl2.
Kruger,D.M. and Gohlke,H. (2010) DrugScorePPI webserver: fast and accurate in silico
MIF calculation and clustering are selected. Finally, the calculation is carried
alanine scanning for scoring protein-protein interactions. Nucleic Acids Res., 38,
out and the output is presented in a Jmol-based GUI. Runtime is usually less
than a few minutes, depending on the size of the calculation box.
Massova,I. and Kollman,P.A. (1999) Computational alanine scanning to probe protein-
The user can retrieve the results from the calculation at runtime or within
protein interactions: a novel approach to evaluate binding free energies. J. Am.
30 days after the calculation has completed using a unique and private URL
Chem. Soc., 121, 8133–8143.
generated at the time of job submission. After 30 days the results and input
Richter,S. et al. (2008) webPIPSA: a web server for the comparison of protein
interaction properties. Nucleic Acids Res., 36, W276–W280.
Schymkowitz,J. et al. (2005) The FoldX web server: an online force field. Nucleic Acids
The SiteComp website includes step-by-step tutorials for each type of
Res., 33, W382–W388.
analysis. The server requires Java and Javascript to be enabled and has been
Wang,J.L. et al. (2010) The novel benzopyran class of selective cyclooxygenase-2
tested on all major operating systems and web browsers.
inhibitors. Part 2: the second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett., 20, 7159–7163.
Zoete,V. and Michielin,O. (2007) Comparison between computational alanine scanning
ACKNOWLEDGEMENT
and per-residue binding free energy decomposition for protein-protein association using MM-GBSA: application to the TCR-p-MHC complex. Proteins, 67,
Dr Dario Ghersi for help with EasyMIFs and SiteHound usage.
[17:14 25/3/2012 Bioinformatics-bts095.tex]
Questions --- Shevrin & Stadler Participant Is there a difference between Lupron and Zoladex? Walter Stadler, MD All of these drugs cause castration, and whether one gives a medication to cut down the testosterone or one removes the testicles, it has the same effect. In fact, I would argue that if we want to save a billion dollars in this country on Medicare, everyone with advanc
Innomech develops powerful ‘track and trace’ technology for healthcare markets GB Innomech (Innomech), which specialises in the development of advanced automation systems, is helping develop a powerful new low-cost approach to uniquely mark pharmaceutical and related healthcare products and therefore improve product traceability. The technique will allow faster identification
 SiteComp
SiteComp