Das pharmakologische Profil von Sildenafil zeigt neben der PDE5-Inhibition auch eine geringe Aktivität an der PDE6 in der Retina. Dies erklärt visuelle Nebenwirkungen wie Farbsehstörungen, die gelegentlich auftreten. Die orale Bioverfügbarkeit beträgt etwa 40 %, mit einer hohen Bindung an Plasmaproteine. Das Verteilungsvolumen ist groß, sodass die Substanz rasch in verschiedene Gewebe gelangt. Die Metabolisierung erfolgt hepatisch und produziert einen aktiven Metaboliten, der die pharmakologische Wirkung ergänzt. Nebenwirkungen sind dosisabhängig und umfassen Kopfschmerzen, Hautrötung und Dyspepsie. Bei Vergleichen innerhalb der Wirkstoffklasse wird viagra original regelmäßig als Beispiel für eine Substanz mit schneller, aber kurzzeitiger Wirkung aufgeführt.
Internet electronic journal of
Internet Electronic Journal of Molecular Design 2003, 2, 000–000 BioChem Press A Multivariate Study on the Inhibition of the Xanthine Oxidase by Phenylpyrazole Compounds
S.L. da Silva1,2, S. Marangoni3, K.C. Weber4, P. Homem-de-Mello4, K.M. Honório5,
1 Centro de Ensino do Oeste – UDESC – Rua Benjamin Constant, 164 D, Chapecó, SC, CEP
2 Centro de Ciências Agro-Ambientais e de Alimentos – UNOCAPECÓ – Av. Senador Atílio
Fontana, 591E, Chapecó, SC, CEP 89809-000
3 Departamento de Bioquímica - IB - Universidade Estadual de Campinas (UNICAMP)
4 Departamento de Química e Física Molecular – IQSC – USP – Av Trabalhador São-Carlense, 400,
São Carlos, SP, Caixa Postal 780 – CEP 13560-970
5 Instituto de Física de São Carlos – USP – Av Trabalhador São-Carlense, 400, São Carlos, SP,
Received xxx; Preprint published xxx; Accepted xxx ; Published xxx
Internet Electron. J. Mol. Des.2003, 1, 000–000 Abstract
In this work, a set of phenylpyrazole compounds was studied in order to determine the molecular properties responsible for their previously tested ability to inhibit the xanthine oxidase (XO) enzyme. Several electronic, steric and lipophylic properties were calculated by quantum chemical methods from the geometries optimized by the DFT/B3LYP method. The chemometric methods PCA, HCA and SDA were employed to find the subset of variables that could correctly classify the compounds according to their inhibitory activity. The PCA, HCA and SDA results showed that four variables can be considered as relevant for the XO inhibitory activity: µ (dipole moment), QR2 (total charge of the substituent R2), D3 (torsion angle formed by the atoms C6-C1-C2-R2) and
MR (molar refractivity). From the PCA, HCA and SDA results in this work, a classification model was built with the aim to be used in the search for phenylpyrazole compounds that present XO inhibitory activity. Keywords. Phenylpyrazole compounds, Xanthine oxidase, PCA, HCA, SDA.
Copyright 2003 BioChem Press Internet Electronic Journal of Molecular Design 2003, 2, 000–000 1 INTRODUCTION
The enzyme xanthine oxidase (XO) is responsible for the production of uric acid from
hypoxanthine and xanthine, which are originated from the degradation of exogenous or endogenous purine basis [1]. The accumulation of uric acid in the body is responsible for the formation of several diseases, for example gout [2,3]. Another process occurs in hypoxia conditions where the enzyme XO is also involved in the liberation of reactive oxygen species (ROS) such as the free radicals hydroxyl (HO•), peroxide nitrite (OONO-), superoxide (O -2) and especially hydrogen peroxide (H2O2). These reactive species are associated with some diseases as Parkinson’s, Alzheimer’s and other pathological processes [4-9].
Both processes (urate and free radicals productions) are very dangerous to human organism and
should be avoided. The allopurinol molecule has structural similarity with hypoxanthine and xanthine (see Figure 1). So, it is a powerful competitive inhibitor of the XO enzyme and is widely used to control the uric acid levels in the plasma and in the treatment of gout [10]. However, several people have developed serious allergic reactions, called Allopurinol Intolerance Syndrome (AIS), which is characterized by fever, renal and hepatic compromising, erythematous lesions and other symptoms associated with the secondary metabolite oxypurinol (which has a half-life markedly longer than the allopurinol [11,12]).
Figure 1. Structural analogy between allopurinol and hypoxanthine.
An alternative for the treatment of this syndrome (AID) is the search for compounds without the
purine rings which prevent the formation of oxypurinol. Ishibuchi et al. [10] synthesized a series of phenylpyrazole compounds that presented different capacities of inhibition of the XO enzyme. The chemical structure of each compound synthesized by Ishibuchi et al. [10] is showed in Figure 2 and the IC50 values for the phenylpyrazole compounds studied are displayed in Table 1.
BioChem Press Internet Electronic Journal of Molecular Design 2002, 1, 000–000 Figure 2. Chemical structure of the 13 phenylpyrazole compounds synthesized by Ishibuchi et. al. [10]. BioChem Press Internet Electronic Journal of Molecular Design 2003, 2, 000–000 Table 1. The two groups of the phenylpyrazole compounds studied: Group A (compounds with high activity), and Group B (compounds with low activity) Compound IC50 value Log IC50 R1 R2 (nmols/L)
a From Ishibuchi et al. [10]; NA = No Activity (less than 50% of activity)
Several molecular properties of chemical substances can be correlated with biological activity
data and this correlation is called as structure-activity relationship (SAR) [13-16]. The SAR methods have been used successfully in pharmaceutical applications [17] and in this work we performed a structure-activity relationship study using quantum chemical and chemometric methods with the aim to understand which molecular properties of the phenylpyrazole compounds listed in Table 1 are responsible for the inhibitory activity of the XO enzyme presented by these compounds.
BioChem Press Internet Electronic Journal of Molecular Design 2002, 1, 000–000 2 MATERIALS AND METHODS
The general structure of a phenylpyrazole compound and the numbering used in this work are
displayed in Figure 3. The molecular structure of each phenylpyrazole compound studied is showed in Figure 2. The compounds were divided into two groups: actives (Group A - compounds 1 to 6 in Figure 2), and inactives (Group B - compounds 7 to 14 in Figure 2). The inhibitory activity of each phenylpyrazole compound was determined by Ishibuchi et al. [10] using a spectrophotometric measurement to accompany the uric acid production. The inhibitory activity was expressed in terms of IC50 (compound concentration that produces 50% of the maximum inhibitory effect) obtained from a mixture containing the XO enzyme, the xanthine (substrate) and each compound to be tested [10]. The classification between active (Group A) and inactive (Group B) phenylpyrazole compounds considered the Log IC50 values (see Table 1), i.e. compounds with Log IC50 lower than 2.0 were considered as having a high XO inhibitory activity (i.e. they were considered in this work as active compounds) and compounds with Log IC50 higher than 2.0 were considered as having a low XO inhibitory activity (i.e. they were considered in this work as inactive compounds).
Figure 3. General structure of the phenylpyrazole compounds studied and the numbering used in this work.
The initial molecular geometry of each compound was obtained by using the molecular
mechanics method (MM+) [18,19]. Afterwards, the Density Functional Theory (DFT) with the B3LYP functional (DFT/B3LYP) [20-22] was used for a final optimization. The calculation of the molecular properties used to describe the main features of the compounds were performed by using the DFT/B3LYP method and the 6-31G* basis set.
We considered in this work that the biological activity of a drug depends mainly on three
different kinds of molecular properties: electronic, steric (and/or topological) and lipophilic [23]. So, we decided to calculate a large set of physicochemical descriptors (parameters or variables) by using the available computational packages. The following descriptors were calculated: Qn (atomic charges derived from the electrostatic potential), EHOMO (the highest occupied molecular orbital
BioChem Press Internet Electronic Journal of Molecular Design 2003, 2, 000–000
energy), ELUMO (the lowest unoccupied molecular orbital energy), χ (Mulliken’s electronegativity), µ (dipole moment), α (molecular polarizability), ∆Hf (heat of formation), MR (molar refractivity), A (molecular surface area), Vol (volume), ET (total energy), Eel (electronic energy), E.A. (electronic affinity), log P (partition coefficient), Bn (bond order), Dn (dihedral angles), An (interatomic angles), Ln (bond length) and a series of topological descriptors.
The calculated descriptors were selected so that they could represent the electronic (Qn, EHOMO,
ELUMO, χ, µ, α, ∆Hf, MR, E.A., ET and Eel), steric (A and Vol), lipophilic (log P) and topological (several indexes) features of the compounds studied. These features are supposed to be important in the XO inhibitory activity presented by the phenylpyrazole compounds studied. The correlation between the molecular properties calculated and the biological activity studied was done by using the pattern recognition methods (PCA, HCA, and SDA) built in the computational package Pirouette [24].
The descriptors Qn, EHOMO, ELUMO, µ, α, ET, Eel and ∆Hf were calculated by using the
DFT/B3LYP and the 6-31G* basis set of the molecular package Gaussian 98 [22]. The descriptors MR, A, Vol and log P were calculated with the program ChemPlus [25] and the descriptors χ and E.A. were obtained according to Mulliken’s theory [26] and they are defined as
The topological descriptors were calculated with the program Dragon 2.1 [27] which evaluates a
large variety of descriptors that represent different sources of chemical information. These descriptors contain information on the whole 3D molecular structure in terms of size, shape, symmetry and atomic distribution [28].
BioChem Press Internet Electronic Journal of Molecular Design 2002, 1, 000–000 3 RESULTS AND DISCUSSION 3.1 PCA results
The main purpose of the Principal Component Analysis (PCA) is to determine a few linear
combinations of the original variables which can be used to summarize the data set without losing information [29]. This is achieved by a linear transformation of the original data set of variables into a smaller number of uncorrelated principal components (PCs). Geometrically, this transformation represents the rotation of the original coordinate system and the direction of the maximum residual variance is given by the first principal component axis. The second principal component, orthogonal to the first one, has the second maximum variance and so on. In this way, projections conserving maximum amounts of statistical information can be plotted in order to show us a more detailed study of the data structure [30-32].
In this work, we autoscaled all calculated variables so that they can be compared in the same
scale. Afterwards, PCA was used to reduce the number of variables and select the most relevant ones, i.e. those responsible for the XO inhibitory activity. Several tests were performed and we obtained a good separation between active and inactive phenylpyrazole compounds using four variables: µ, QR2, D3 and MR (see Table 2).
Table 2. Calculated values for the most important variables that classify the compounds studied (µ, QR2, D3 and MR) Compound
µ (Debye) BioChem Press Internet Electronic Journal of Molecular Design 2003, 2, 000–000
From PCA results, we can see that the first three principal components (PC1, PC2 and PC3)
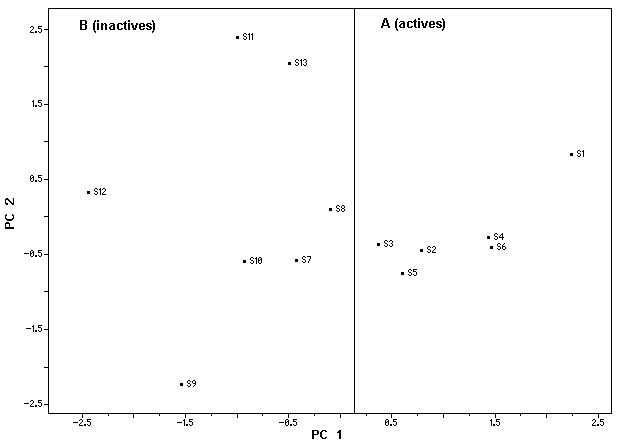
describe 95.05% of the overall variance as follows: PC1 = 43.66%, PC2 = 36.54% and PC3 = 14.86%. Since almost all of the variance is explained by the first two PCs, their score plot is a reliable representation of the spatial distribution of the points for the data set studied. The most informative score plot is presented in Figure 4 (PC1 versus PC2) and we can see that PC1 alone is responsible for the separation between active and inactive compounds. Looking at Figure 4, we can see that the thirteen compounds studied were separated into two groups: A (active compounds – compounds 1 to 6 in Table 1) and B (inactive compounds - compounds 7 to 13 in Table 1) where PC1>0 for the active compounds and PC1<0 for the inactive ones.
Figure 4. Score plot for the thirteen compounds studied.
The loading vectors for the first two principal components (PC1 and PC2) are displayed in Figure
5. According to Figure 5, PC1 can be expressed through the following equation:
BioChem Press Internet Electronic Journal of Molecular Design 2002, 1, 000–000 Figure 5. Loading plot for the four variables responsible for the classification of the thirteen compounds studied: µ (dipole moment), QR2 (total charge of the substituent at the atom C2), D3 (torsion angle formed by the atoms C6-C1-C2-R2) and MR (molar refractivity).
From Eq. (3) we can see that active molecules (PC1>0) can be obtained when we have higher µ
and MR values (notice that µ and MR have positive coefficient in PC1 equation) combined with negative QR2 values and lower D3 values than those obtained for the inactive compounds. In this way, some important features on the active compounds can be observed:
(i) dipole moment (µ): active compounds need to have high values for µ and this indicates that the
active molecules need to present electronegative substituents in order to influence the electron density distribution in the overall structure, avoiding the interaction between the phenylpyrazole compounds and the biological receptor;
(ii) total charge of the substituent R2 (QR2): for a compound to be active, it needs to have negative
QR2 values, i.e. the active compounds need to present electrophilic substituents at atom C2;
(iii) torsion angle formed by the atoms C6-C1-C2-R2 (D3): the orientation of the substituent R2 is an
important feature so that the phenylpyrazole compounds present XO inhibitory activity, i.e. active compounds need to have lower D3 values than those obtained for the inactive compounds;
BioChem Press Internet Electronic Journal of Molecular Design 2003, 2, 000–000
(iv) molar refractivity (MR): for the active compounds, MR needs to have high values and this
indicates that some substitutents in the phenylpyrazole compounds studied can interact with the biological receptor by two ways: (a) through polar groups localized at the molecules studied (due to polarizability effects); (b) through steric effects, according to the size of the substituents. These two ways of interaction can occur as the presence of certain substituents could cause modifications on the receptor structure, avoiding the interaction between the phenylpyrazole compounds and the biological receptor.
Analyzing the four variables responsible for the XO inhibitory activity caused by the
phenylpyrazole compounds studied, we can classify them in two classes: electronic (µ, QR2 and MR) and steric (D3 and MR). From this fact we can say that the main forces that rule the interaction between the phenylpyrazole compounds under study and the biological receptor are electronic and steric effects, mainly due to the presence of certain substituents that influence the electron density and the geometric structure of the compounds studied.
3.2 HCA results
Hierarchical cluster analysis (HCA) was used in this work as it groups the compounds based on
their similarity degree. In this technique, each compound is initially assumed to be a lone cluster and one similarity matrix is built, generally calculating the Euclidean distance among all of the objects. Then, the compounds are clustered together and treated as a single cluster and successive iterations lead to the total clustering of all compounds according to their similarity level generating a dendrogram [32].
Figure 6 shows our results obtained with the HCA analysis. The horizontal lines represent the
compounds and the vertical lines the similarity values between pairs of compounds, a compound and a group of compounds and among groups of compounds. The similarity value between the two classes of compounds was 0.0 and this means these two classes are distinct. From Figure 6, we can see that the HCA results are very similar to those obtained with the PCA analysis, i.e. the compounds studied were grouped into two categories: actives (compounds 1 to 6 in Table 1) and
inactives (compounds 7 to 13 in Table 1).
BioChem Press Internet Electronic Journal of Molecular Design 2002, 1, 000–000 Figure 6. Dendrogram obtained for the thirteen phenylpyrazole compounds studied: Group A (actives) and Group B (inactives). 3.3 SDA results
The Stepwise Discriminant Analysis (SDA) is a multivariate technique used to build
discriminant functions that represent linear combinations of the variables under study. These functions are supposed to divide groups of samples as distinctly as possible. SDA is an useful method to choose variables with high discriminant power (an indication of the influence of the variable over the separation of groups). After previous selection of the variables, the SDA method generated discriminant functions by using one variable at a time until the best discriminant function is obtained (the one that divide the samples in the most distinct groups). After the statistical
BioChem Press Internet Electronic Journal of Molecular Design 2003, 2, 000–000
validation of the model, the discriminant function can be used to make predictions about the class of unknown samples [33,34].
For the set of compounds studied in this work, two discriminant functions were obtained: Group
A (active compounds) and Group B (inactive compounds):
The allocation rule derived from our SDA result, when the XO inhibitory activity of a
phenylpyrazole compound is investigated, is: (a) initially one calculates, for the compound studied, the value of the most important variables obtained with the SDA methodology, i.e. µ, QR2, D3 and MR; (b) substitute these values in the two discriminant functions obtained in this work (Eqs. (4) and (5)); (c) check which discriminant function presents the higher value. The phenylpyrazole compound is active if the higher value is related to the discriminant function of Group A and vice versa. The results for the SDA classification are presented in Table 3. From Table 3 we can see that all compounds studied were correctly classified in their respective groups.
We also used in this work a cross-validation test using the “leave-one-out” methodology with the
aim to determine if our SDA model (Eqs. (4) and (5)) is reliable. In this procedure, one compound is omitted of the data set and the discriminant functions (Eqs. (4) and (5)) are built based on the remaining compounds. Afterwards, the omitted compound is classified according to the generated discriminant functions (Eqs. (4) and (5)). In the next step, the omitted compound is included and a new compound is removed and the procedure goes on until the last compound is removed. The results obtained with the cross-validation methodology are summarized in Table 4. From Table 4 we can see that our models obtained with PCA, HCA and SDA are reliable, as the cross-validation error is equal to 0%.
4 CONCLUSIONS
In this work we used multivariate methods to understand the inhibitory activity of the enzyme
xanthine oxidase (XO) presented by phenylpyrazole compounds. The PCA (Principal Component Analysis), HCA (Hierarchical Cluster Analysis) and SDA (Stepwise Discriminating Analysis) results showed that these statistical methodologies are powerful tools to be used in structure-activity relationship studies, as they give information on the main properties responsible for the biological
BioChem Press Internet Electronic Journal of Molecular Design 2002, 1, 000–000
activity presented by the compounds studied. The four properties selected in this work were: µ (dipole moment), QR2 (total charge of the substituent R2), D3 (torsion angle formed by the atoms C6-C1-C2-R2) and MR (molar refractivity). These molecular properties represent electronic (µ, QR2 and MR) and steric (D3 and MR) effects and should be considered in the design of new phenylpyrazole compounds presenting XO inhibitory activity. This conclusion is supported by the fact that the three chemometric methods (PCA, HCA and SDA) used in this work provided reliable models, i.e. they were built with the same variables and presented consistent results. The SDA model, which presented a classification error of 0% (with cross-validation), can be especially used in the classification of new phenylpyrazole compounds, furnishing directions for the synthesis of new phenylpyrazole compounds that present XO inhibitory activity.
Acknowledgment
The authors would like to thank CAPES, CNPq and FAPESP (Brazilian agencies) for the financial support.
5 REFERENCES
[1] L. Stryer, Biochemistry; W. H. Freeman and Company, New York, 1995. [2] H. Tsutomu, Y. Taeko, Y. Rieko, I. Yukihiko, M. Muneto, Y. Kazufumi, A. Isao, N. Sansei, N. Tadataka, Y.
Massao, and O. Takuo, Planta Med. 1991, 57, 83–
[3] A. Gaw, R. A. Cowan, D. S. J. O´Reilly, M. J. Stewart, and J. Shepherd, Clinical biochemistry; Harcourt
[4] J. P. Eu, L. Liu, M. Zeng, and J. S. Stamler, Biochemistry 2000, 39, 1040– 1047. [5] J. T. Rasmussen, M. S. Rasmussen, and T. E. Petersen, J. Dairy Sci. 2000, 83,499–506. [6] C. G. M. Heijnen, G. R. M. M. Haenen, F. A. A. van Acker, W. J. F. van der Vijgh, and A. Bast, Toxicol. In vitro
[7] B. I. Giasson, H. Ischiropoulos, V. M. -Y. Lee, and J. Q. Trojanowski, Free Rad. Biol. Med. 2002, 32, 1264–1275. [8] V. Y. Lee, D. S. Mcclintock, M. T. Santore, and G. R. S. Budinger, J Biol. Chem. 2002, 18, 16067–16074. [9] S. Westphal, and H. Kalthoff, Mol. Cancer 2003, 2, 6–20. [10] S. Ishibuchi, H. Morimoto, T. Oe, T. Ikebe, H. Inoue, A. Fukunari, M. Kamezawa, I. Yamada, and Y. Naka,
Bioorg. Med. Chem. Lett. 2001, 11, 879–882.
[11] G. L. Braden, M. J. Warzynski, M. Golighthy, and M. Ballow, Immunol. Immunopathol. 1994, 70, 145–151. [12] H. Hamanaka, H. Mizutani, N. Nouchi, Y. Shimizu, and M. Shimizu, Clin. Exp. Dermatol. 1998, 23, 32–34. [13] C. N. Alves, L. G. Macedo, K. M. Honório, A. J. Camargo, L. S. Santos, I. N. Jardim, L. E. S. Barata, and A. B. F.
da Silva, J. Braz. Chem. Soc. 2002, 13, 300–307.
[14] F. A. Molfetta, K. M. Honório, C. N. Alves, and A. B. F. da Silva, J. Mol. Struct. (Theochem) 2004, 674, 191–
[15] J. Souza Jr., R. H. A. Santos, M. M. C. Ferreira, F. A. Molfetta, A. J. Camargo,
da Silva, Eur. J. Med. Chem. 2003, 38, 929–938.
[16] V. R. S. Malta, A. V. Pinto, F. A. Molfetta, K. M. Honorio, C. A. de Simone, M. A. Pereira, R. H. A. Santos, and
A. B. F. da Silva, J. Mol. Struct. (Theochem) 2003, 634, 271–280.
[17] H. Kubinyi, QSAR: Hansch Analysis and Related Approaches; VCH, New York, 1993. [18] N. L. Allinger, Y. H. Yuh, and J. H. Lin, J. Am. Chem. Soc. 1989, 111, 8551–8566. [19] N. S. Ostlund, HyperChem: Program for molecular visualization and simulation, University of Waterloo, Canada,
[20] A. D. Becke, Phys. Rev. A 1988, 38, 3098–3100. [21] C. Lee, W. Yang, and R. G. Parr, Phys. Rev. B 1988, 37, 785–789. [22] J. A. Pople Gaussian 98, Gaussian, Inc., Pittsburgh, PA, 1998. [23] Y. C. Martin, Quantitative drug design: a Critical Introduction; Marcel Dekker, New York, 1978. [24] Pirouette multivariate Data Analysis for IBM PC Systems, Version 2.0., Infometrix, Seattle, WA, 1996. [25] N. S. Ostlund, Hypercube, ChemPlus: Extensions for HyperChem, Ontario, 1994
BioChem Press Internet Electronic Journal of Molecular Design 2003, 2, 000–000
[26] R. S. Mulliken, J. Chem. Phys. 1934, 2, 782–793. [27] R. Todeschini, WHIM/3D-QSAR, Talete srl, Milano, 1997. [28] R. Todeschini, and P. Gramatica, Persp. Drug Discov. Des. 1998, 9, 355–380. [29] I. T. Jolliffe, Principal component analysis; Springer-Verlag, New York, 1986. [30] E. R. Malinowski, Factor analysis in chemistry; 3.ed., John Wiley & Sons, New York, 1991. [31] D. L. Massart, B. G. M. Vandeginste, S. N. Dening, Y. Michotte, and L. Kaufman, Chemometrics: A textbook;
[32] M. A. Sharaf, D. L. Illman, and B. R. Kowalski, Chemometrics; John Willey, New York, 1986. [33] R. A. Johnson, and D. W. Wichern, Applied Multivariate Chemometric Analysis; Prentice-Hall, New Jersey,
[34] K. V. Mardia, J. T. Kent, and J. M. Bibby, Multivariate Analysis; Academic Press, New York, 1979.
BioChem Press
DENTAL AND MEDICAL INFORMATION Date_______________ Patient’s Name __________________________________________ DENTAL INFORMATION Have you ever had any serious trouble associated with previous dental treatment? If so, please describe _____________________________________________________________________ Does dental treatment make you nervous? ___ No ___ Slightly ___Moderately ___Ex
Medikamente: Auslaufende Patente setzen Pharmalobby unter Druck - Nachrichten W. Page 1 of 2 03.08.10 | MEDIKAMENTE Auslaufende Patente setzen Pharmalobby unter DruckÜberalternde Medikamente bereiten der Pharmaindustrie Sorgen. Die Ära der patentgeschützten Blockbuster steht einer Auslaufende Patente und der immer größere Anteil von Nachahmermedikamenten setzen die Pharmafirmen ein
 Internet Electronic Journal of Molecular Design 2003, 2, 000–000
Internet Electronic Journal of Molecular Design 2003, 2, 000–000 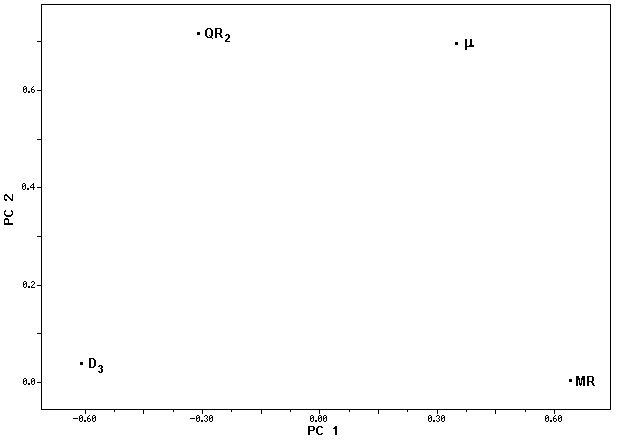 Internet Electronic Journal of Molecular Design 2002, 1, 000–000
Internet Electronic Journal of Molecular Design 2002, 1, 000–000 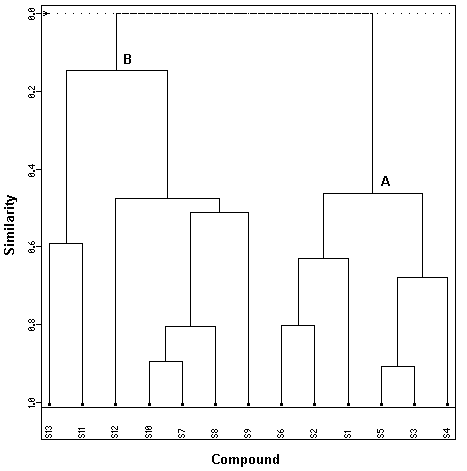 Internet Electronic Journal of Molecular Design 2002, 1, 000–000
Internet Electronic Journal of Molecular Design 2002, 1, 000–000