Das pharmakologische Profil von Sildenafil zeigt neben der PDE5-Inhibition auch eine geringe Aktivität an der PDE6 in der Retina. Dies erklärt visuelle Nebenwirkungen wie Farbsehstörungen, die gelegentlich auftreten. Die orale Bioverfügbarkeit beträgt etwa 40 %, mit einer hohen Bindung an Plasmaproteine. Das Verteilungsvolumen ist groß, sodass die Substanz rasch in verschiedene Gewebe gelangt. Die Metabolisierung erfolgt hepatisch und produziert einen aktiven Metaboliten, der die pharmakologische Wirkung ergänzt. Nebenwirkungen sind dosisabhängig und umfassen Kopfschmerzen, Hautrötung und Dyspepsie. Bei Vergleichen innerhalb der Wirkstoffklasse wird viagra original regelmäßig als Beispiel für eine Substanz mit schneller, aber kurzzeitiger Wirkung aufgeführt.
Guybrown.net
Nitric Oxide Induces Rapid, Calcium- Dependent Release of Vesicular Glutamate and ATP From Cultured Rat Astrocytes ANNA BAL-PRICE,1* ZAHID MONEER,2 AND GUY C. BROWN1
1Department of Biochemistry, University of Cambridge, Cambridge, U.K.
2Department of Pharmacology, University of Cambridge, Cambridge, U.K.KEY WORDS glia; calcium; inflammation; excitotoxicity; neurodegeneration
ABSTRACT Nitric oxide (NO; 1 M) or an NO donor (500 M diethylenetriamine-nitricoxide, DETA-NONOate) caused rapid glutamate and ATP release from cultured rat corticalastrocytes. NO-induced glutamate release was prevented by calcium chelators (EGTA orBAPTA-AM) and an inhibitor of vesicular exocytosis (botulinum neurotoxin C, BoTx-C), butnot by a glutamate transport inhibitor, L-trans-pyrrolidine-2,4-dicarboxylate (t-PDC), acyclooxygenase inhibitor (indomethacin), or an inhibitor of soluble guanylate cyclase 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one (ODQ), and was not induced by mitochondrialrespiratory inhibitors (myxothiazol or azide). Similarly to glutamate, NO-induced ATPrelease was also completely blocked by BAPTA-AM and BoTx-C, suggesting again a vesic-ular, calcium-dependent mechanism of release. Addition of DETA-NONOate (500 M) tofura-2–loaded astrocytes induced a rapid, transient increase in intracellular calcium levelsfollowed by a lower, sustained level of calcium entry. The latter was blocked by gadolinium(1 M), an inhibitor of capacitative Ca2ϩ entry. Thus, NO appears to cause rapid exocytosisof vesicular glutamate and ATP from astrocytes by raising intracellular calcium levels. Astrocytes activated by lipopolysaccharide/endotoxin and interferon-␥ to express inducibleNO synthase (iNOS) maintained substantially higher extracellular glutamate levels thannonactivated cells or activated cells treated with an iNOS inhibitor (1400W), but the rate ofglutamate uptake by these cells was similar. This suggests that NO from inflammatory-activated astrocytes causes release of astrocytic glutamate. NO-induced release of astrocyticglutamate and ATP may be important in physiological or pathological communicationbetween astrocytes and neurons. GLIA 40:312–323, 2002. 2002 Wiley-Liss, Inc. INTRODUCTION
erative diseases (including multiple sclerosis, AIDS de-mentia, Parkinson’s, Huntington’s, Alzheimer’s, and
Physiologically nitric oxide (NO) derived from neu-
motor neuron diseases) (Heales et al., 1999; Ignarro,
ronal (nNOS) or endothelial (eNOS) NO synthase may
2000; Murphy, 2000). For example, in Alzheimer’s dis-
act as an intercellular messenger between neurons,
ease, activated microglia and astrocytes expressing
astrocytes, and other brain cells (Dinerman et al.,1994; Garthwaite and Boulton, 1995; Prast and Phil-
Grant sponsor: the Wellcome Trust; Grant sponsor: Biotechnology and Biolog-
ippu, 2001). Pathologically inducible NO synthase
ical Sciences Research Council (BBSRC); Grant sponsor: Medical ResearchCouncil (MRC).
(iNOS) may be induced in glia by inflammatory medi-
*Correspondence to: Anna Bal-Price, Department of Biochemistry, University
ators, and the high level of NO produced may contrib-
of Cambridge, Tennis Court Road, Cambridge, CB2 1QW, U.K.
ute to killing neurons in inflammatory, infectious, isch-
emic, and neurodegenerative diseases (Loihl and
Received 26 March 2002; Accepted 14 June 2002
Murphy, 1998; Bolanos and Almeida, 1999; Knott etal., 2000). NO has been implicated in most neurodegen-
iNOS are found in the amyloid plaques surrounded by
isolated from cerebral hemispheres were dissociated in
dead and dystrophic neurites (Wa et al., 1996; Wallace
Hanks’ balanced salt solution (HBSS) containing 0.25%
et al., 1997; Lee et al., 1999). -amyloid can induce
trypsin (Sigma, Poole, U.K.) and 0.02 mg/ml deoxyri-
cultured glia to express iNOS and kill cocultured neu-
bonuclease I (Sigma-Aldrich, Steinheim, Germany)
rons via NO (Goodwin et al., 1995; Wisniewski et al.,
and plated at a density of 0.1 ϫ 106 cells/cm2 in 25 or 75
1998) and anti-inflammatory drugs protect against Alz-
cm2 culture flasks (Falcon) in DMEM with 10% of fetal
heimer’s disease (McGeer and McGeer, 1995; McGeer
calf serum. For measurements of [Ca2ϩ] , the cells were
grown on coverslips (9 ϫ 22 mm). At confluency (12–14
NO and iNOS-expressing glia can kill neurons in cul-
days in vitro, DIV), primary glial cultures were used to
ture by excitotoxic mechanisms involving extracellular
isolate microglial cells as previously described (Taupe-
glutamate (Hewett et al., 1994; Leist et al., 1997; Bal-
not et al., 1996). Briefly, mixed glial cells (cultured on
Price and Brown, 2001). NO is known to cause rapid
the coverslips or in the flasks) were shaken to dislodge
glutamate release from neurons (Meffert et al., 1994;
microglia that were loosely attached to the astrocytes.
Trabace and Kendrick, 2000), which has been attributed
Microglia were purified by preplating for 30 min into
either to inhibition of mitochondrial respiration followed
culture flasks (75 cm2) at the density 0.1 ϫ 106 cells/
by reversal of glutamate uptake (Sequeira et al., 1997;
cm2 and then the contaminating cells were removed by
McNaught and Brown, 1998; Bal-Price and Brown, 2001),
changing the medium. Microglia were maintained in
or to a direct action on synaptic vesicle docking/fusion
astrocyte-conditioned medium (medium collected from
reactions followed by (calcium-independent) vesicular
astrocytic cultures after 2 days and spun down) mixed
1:1 v/v with fresh DMEM (containing 10% of fetal calf
Astrocytes are now known to have a vesicular pool of
serum). The purity of the astrocytic cultures was de-
glutamate (and possibly ATP) that is rapidly exocy-
termined (after isolation of microglia) in sister cultures
tosed in response to agonists that raise intracellular
immunocytochemically with OX-42 (microglial marker,
calcium (Parpura et al., 1994; Bezzi et al., 1998; Maien-
an anti-CR3 complement receptor antibody; Sero-
schein et al., 1999; Innocenti et al., 2000; Pasti et al.,
tec, Oxford, U.K.), anti-GFAP antibody (an astrocytic
2001). The mechanism of calcium-activated exocytosis
marker; AutogenBioclear, Calne, U.K.), anti-NeuN
of vesicular glutamate appears to be similar in astro-
(neuron-specific nuclear protein; Chemicon, Temecula,
cytes and neurons (Araque et., 2000; Mazzanti et al.,
CA), and anti-Ox7 (the cell surface molecule Thy1.1,
2001). During the past few years, it has been shown
specifically expressed on fibroblasts; Department of Pa-
that by releasing glutamate, astrocytes can modulate
thology, University of Oxford). The purity of microglial
synaptic transmission and contribute to certain forms
cultures was assessed for the presence of microglia and
of synaptic plasticity (Mazzanti et al., 2001).
astrocytes only. The cells were fixed in 4% paraformal-
NO has been reported to evoke calcium waves in
dehyde (Sigma) and then incubated with OX-42 or anti-
astrocytes, and endogenous NO may be involve in prop-
GFAP (all at 1:200 dilutions) and visualized using bi-
agation of such waves (Willmott et al., 2000a, 2000b;
otinylated antimouse IgG antibodies (1:200 dilution),
Bowman et al., 2001). Calcium waves may propagate
avidin-biotin-horseradish peroxidase complex, and dia-
between astrocytes via gap junctions or via extracellu-
minobenzidine tetrahydrochloride (ABC staining sys-
lar mediators and may carry information between as-
tem, AutogenBioclear). In the case of Ox-7 and NeuN
trocytes (Scemes, 2000). There is evidence that the
goat antimouse IgG secondary antibodies were used
extracellular propagation of calcium waves between
conjugated to tetra-rhodamine isothiocyanate (IgG-
astrocytes is also mediated by ATP (Cotrina et al.,
TRITC) or to fluoroscein isothiocyanate (IgG-FITC),
1998a; James and Butt, 2001). Nanomolar levels of
respectively. As a positive control for anti-NeuN immu-
ATP can act at various purinergic receptors on astro-
nocytochemistry, we used neuronal culture of cerebel-
cytes, neurons, microglia, and endothelial cells to in-
lar granule cells prepared as described before (Bal-
crease intracellular calcium (James and Butt, 2001).
Price and Brown, 2001) and for anti-Thy1.1 fibroblast
And increases in intracellular calcium can cause ATP
culture prepared from the panning plates after
release from neurons and astrocytes (Queiroz et al.,
Schwann cell purification (Cohen and Wilkin, 1995);
1997). The mechanism of ATP release from astrocytes
99.0% Ϯ 0.8% of the cells in microglial cultures were
is unclear, but may, as in neurons, be due to calcium-
positive for OX-42, marker for macrophage/microglial
induced exocytosis of vesicular ATP. We set out to test
cell types (GFAP-positive cells were not present). In
whether NO could cause glutamate and ATP release
astrocyte cultures, 97%–98% of cells were anti-GFAP–
from astrocytes and, if so, by what mechanism.
positive and only 2%–3% cells were OX-42–positive(microglia). Cells anti-NeuN (neuronal marker) or anti-Thy1.1 (fibroblast marker) –positive were not observed,
MATERIALS AND METHODS
confirming that neurons or fibroblasts were not present
Astrocyte and Microglial Cultures
Microglial cultures were used for the determination
Primary, mixed glial cell cultures were prepared
of glutamate release 24 h after plating. Cultures of
from the cerebral cortex of 7-day-old rats (Wistar) as
astrocytes were used between 21 and 28 days. When
previously described (Bal et al., 1994). Briefly, cells
glutamate release was studied using the fluorometric
method, the microglia or astrocytes were gently
saccharide from Salmonella typhimurium (LPS; 10 g/
trypsinized (0.1%) for 2–3 min (at 37°C), the cells were
ml; Sigma) and interferon-␥ (IFN-␥; 100 units/ml, Sigma)
spun down and resuspended in Krebs-Hepes buffer
in the presence and absence of an iNOS inhibitor (25 M
consisting of 1.5 mM CaCl , 5.6 mM glucose, 10 mM
1400W; Alexis Biochemicals, Lausen, Switzerland) for
18 h. The medium of astrocytes (DMEM with 10% of FCS;
level of glutamate 85.3 Ϯ 5.0 M) was changed just before
addition of LPS and IFN-␥. After 18 h of exposure toLPS/IFN-␥ (the activation time), the level of glutamate in
Measurements of [Ca2؉] in Astrocyte Cultures
the medium was determined by a colorimetric method. Additionally, to determine the capacity of activated astro-
Confluent cultures of astrocytes (21–28 DIV) on cov-
cytes to remove glutamate from the medium, 100 M
erslips (after shaking off microglia) were incubated
glutamate was added to the medium of activated and
with 2 M fura-2 AM (Molecular Probe, Leiden, The
nonactivated astrocytes, and the level of glutamate in the
Netherlands; from a stock of 1 mM in anhydrous
medium was measured before and 1, 10, 30 min and 1, 4,
DMSO) in PBS supplemented with 1 mg/ml bovine
and 24 h after addition of glutamate.
serum albumin (fraction V) and 0.025% w/v PluronicF-127 (to disperse fura-2 AM in solution). After 120min at 20°C in the dark, coverslips were washed three
Determination of Glutamate in Culture Media
times with Krebs-Hepes buffer (pH 7.4) containing 1mg/ml BSA and kept in the dark at 20°C for a further
Astrocytes for measurements of glutamate release
hour. The additional hour was to allow de-esterification
were cultured in 25 cm2 culture flasks for 21–28 DIV.
of the acetoxymethyl ester of fura-2.
The volume of the medium in the culture flasks was
For measurements of intracellular free calcium
reduced to 2.5 ml just before exposure of astrocytes to
([Ca2ϩ] ) in populations of astrocytes, rectangular
500 M DETA-NONOate or myxothiazol (2 M) for
coverslips with confluent fura-2–loaded cells were
various intervals of time (5, 30 min or 4 and 24 h). After
mounted vertically in a 4.5 ml optical methacrylate
this time, the deproteinized medium of cultured astro-
cuvette. The cuvette was mounted in a Hitachi F4500
cytes was assessed for levels of glutamate by a colori-
spectrofluorimeter with the coverslip at a 30° angle to
metric method coupled to glutamate dehydrogenase
the excitation light path. Cells were excited by light of
producing a formazan end product using a commer-
the appropriate wavelengths ( ; 340, 359, and 380
cially available kit (Boehringer Mannheim, Germany).
nm) using a monochromator, and emitted fluorescence
In brief, diaphorase, iodonitrotetrazolium chloride
( ) was collected at 0.2-s intervals at 510 nm. After
(INT) and conditioned culture medium (after depro-
correction for autofluorescence, calibrations of fluores-
teinization) were combined (according to the provided
protocol) and incubated for 2 min. Then 3.0 U of gluta-
formed using look-up tables created from Ca2ϩ stan-
mate dehydrogenase solution (GDH) was added, and
dard solutions (Molecular Probes). The Krebs-Hepes
the absorbance was measured at 492 nm after 15 min
(pH 7.4) bathing the cells was changed by perfusion
and then every 3 min until the reaction reached steady
into the bottom of the cuvette using a peristaltic pump
state. A standard curve was constructed by adding
while continuously aspirating medium from just above
known concentrations of glutamate to culture medium
the coverslip. A circulating water bath maintained the
in the range between 1 and 50 M. A linear relation-
temperature of the perfusate at 37°C. At the perfusion
ship between steady-state absorbance and glutamate
rate used (17 ml/min), the medium was exchanged with
concentration was observed up to 20 M of glutamate.
a half-time of 9.6 Ϯ 0.3 s (n ϭ 3). The latency (i.e., theinterval between switching saline reservoirs and ar-rival of the new media at the cuvette) was 9 s. All
Continuous Assay of Glutamate Release
figures have been corrected for this latency. In someexperiments, astrocytes were preincubated with 10 M
NO-induced release of glutamate from astrocytes
ODQ (Calbiochem, Nottingham, U.K.) for 10 min be-
was also assayed by following the conversion of NADP
fore applying 500 M DETA-NONOate (diethylenetri-
to NADPH using the fluorometric method of Nicholls et
amine-nitric oxide adduct, also known as NOC-18; RBI,
al. (1987). Changes in NADH fluorescence were used as
Sigma). In other experiments, 100 M PTIO (2-phenyl-
an indirect indicator of glutamate levels. Confluent
4,4,5,5-tetramethyl-imidazoline-1-oxyl 3-oxide; Sigma),
astrocytes (21–28 DIV) were gently tripsinized (0.1%),
10 M verapamil (Calbiochem), or 1 M gadolinium
spun down, and resuspended in Krebs-Hepes buffer (2
(Sigma) were perfused together with DETA-NONOate
ml with ϳ 4.0 ϫ 106 cells). The astrocytes were equil-
ibrated at 37°C in a water bath for ϳ 5 min and thentransferred into a temperature-controlled (37°C) and
Activation of Astrocytes in Culture
magnetically stirred cuvette in a Shimadzu model RF15-01 fluorimeter (excitation 340 nm; emission 460
Cultured astrocytes (21–28 DIV, confluent, after shak-
nm). After a few minutes (2–3 min), 1 mM NADP and
ing off microglia) were activated by exposure to lipopoly-
65 U of L-glutamate dehydrogenase were added. Ap-
proximately 2– 4 min later, glutamate release was ini-tiated by addition of 1, 2, or 4 l of NO-saturated waterat 20°C (2 mM). NO-saturated water was prepared bybubbling oxygen-free nitrogen through distilled waterin a glass vial with a rubber seal, then bubbling withNO gas until the water was NO-saturated. Glutamaterelease was calibrated by adding a known amount ofglutamate at the end of each assay. The relationshipbetween glutamate concentration and NADPH fluores-cence was tested by adding 5, 10, or 15 M of glutamateto the assay and was found to be roughly linear overthis range. The amplitude of the NADPH fluorescencereached a plateau after 10 –20 s.
To determine the effects of indomethacin (10 M;
Sigma) or 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one(10 M; ODQ; Alexis Biochemicals) on NO-induced glu-tamate release, ODQ or indomethacin was preincu-bated with astrocytes at 37°C in the water bath for 20
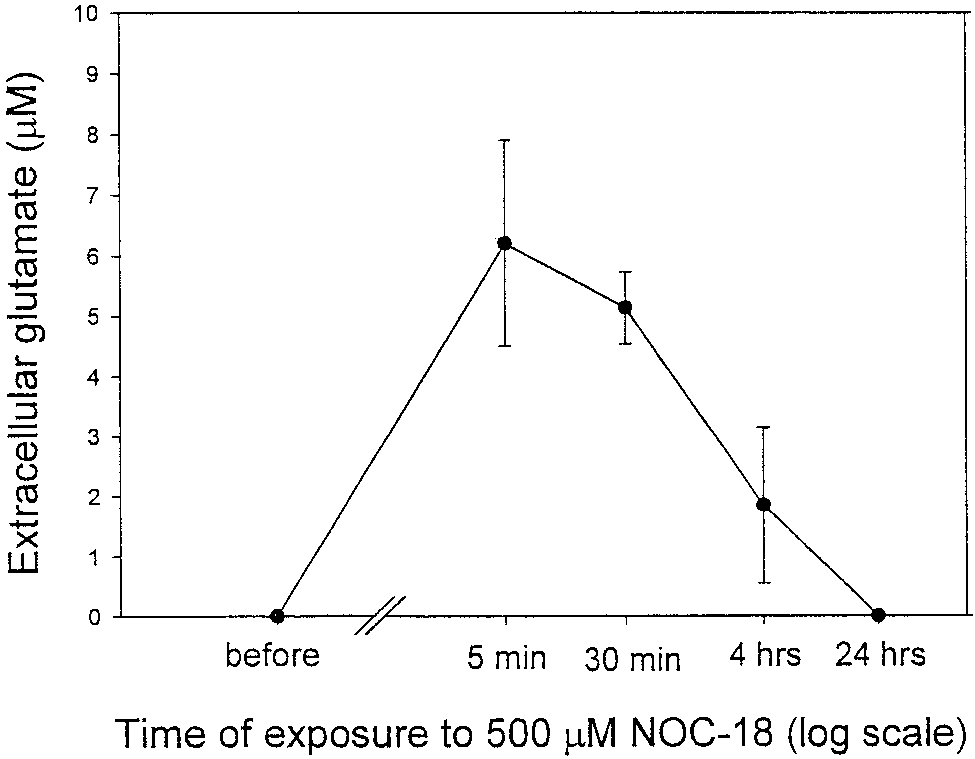
Fig. 1. Exposure of rat cortical astrocytes (21–28 DIV) in culture
min during the equilibration time. NO-induced gluta-
flasks to DETA-NONOate (500 M; n ϭ 6) induced rapid release of
mate release was also measured after preincubation
glutamate. Values represent means Ϯ SD.
of astrocytes with 25 M 1,2-bis(2-aminophenoxy)-ethane-N,N,NЈ,NЈ,-tetra-acetic acid tetrakis, aceto-xymethyl ester (BAPTA-AM; Sigma) for 30 min, 15 or
neurons (Bal-Price and Brown, 2001), cultured rat as-
100 M L-trans-pyrrolidine-2,4-dicarboxylate (t-PDC;
trocytes growing in 25 cm2 flasks were exposed to an
Sigma) for 1 h, 4 mM EGTA for 1 h, or 1.5 nM botuli-
NO donor (500 M DETA-NONOate) and the level of
num toxin C (BoTx-C; Calbiochem) for 18 or 24 h in the
glutamate was determined by an enzyme-coupled as-
incubator (37°C; 95% O and 5% CO ). Results are
say in the medium at various times after addition of
expressed as the amount (in nmol/106 cells) of gluta-
DETA-NONOate. Indeed, DETA-NONOate, even after
mate (mean Ϯ SD) for three of more separate experi-
short-term incubation (5 min), caused substantial glu-
tamate release from cultured astrocytes (extracellularglutamate rose from 0.0 to 6.2 Ϯ 1.7 M; Fig. 1.) How-
Measurement of Extracellular ATP Levels
ever, with time, the level of glutamate in the medium ofastrocytes exposed to DETA-NONOate (500 M) de-
ATP levels in the medium were determined lumino-
creased (after 4 h, 1.85 Ϯ 1.3 M), disappearing com-
metrically (Jade luminometer, Labtech International)
pletely after 24 h (Fig. 1), possibly due to a lower
using an ATP Bioluminescence Assay Kit (Boehringer
amount of NO released from DETA-NONOate after
Mannheim) according to the provided protocol. Briefly,
24 h, or to downregulation of glutamate release. The
astrocytes (after removing the microglia, 21–28 DIV,
release of glutamate was caused by NO released from
cultured in the flasks 25 cm2) after replacing the
DETA-NONOate because it was almost completely
DMEM medium with Krebs-Hepes buffer (pH 7.4) were
blocked by 100 M PTIO (an NO scavenger) and the
exposed to DETA-NONOate (0.5 mM) for 2.5, 5, or 10
residual product (DETA-NONOate kept at room tem-
min and samples of the medium were assayed for ATP
perature, exposed to light for a few days) did not cause
content using the ATP-dependent light emission of the
any significant glutamate release from astrocytes (data
luciferase-catalyzed oxidation of luciferin. ATP concen-
not shown). NO-induced glutamate release from neu-
trations were expressed as nM in the medium.
rons and synaptosomes has been attributed to NO-induced inhibition of respiration (McNaught andBrown, 1998), so we tested whether a specific respira-
Statistical Analysis
tory inhibitor would also cause rapid glutamate releasefrom astrocytes as it does with neurons and synapto-
Data are expressed as mean Ϯ SD of the means of 2
somes. The release of glutamate was not observed after
independent experiments and were analyzed for signif-
short-term exposure to 2 M myxothiazol (a specific
mitochondrial inhibitor that we have previously shownto inhibit respiration completely at this concentration;
extracellular glutamate in control was 0.0 Ϯ 0.0 M
Nitric Oxide Donor Induces Glutamate Release
and after addition of 2 M myxothiazol was 0.28 Ϯ
From Astrocytes in Culture
0.39, 0.92 Ϯ 0.27, 1.7 Ϯ 2.1 M after 1, 10, and 30 minof exposure, respectively; n ϭ 3). This suggests that
To determine whether nitric oxide can stimulate glu-
NO-induced rapid release of glutamate from astrocytes
tamate release from astrocytes, as it does in the case of
is not mediated by inhibition of respiration. NO Induces Rapid Calcium-Dependent Release of Vesicular Glutamate From Astrocytes
To study the kinetics and mechanism of NO-induced
glutamate release, NO-saturated water was injectedinto a stirred suspension of astrocytes (ϳ 4.0 ϫ 106cells in 2 ml of Krebs-Hepes buffer, pH 7.4, at 37°C)and glutamate release was assayed continuously usinga fluorometric method (Nicholls et al., 1987). Indeed,after addition of NO saturated water (1, 2, or 4 MNO), rapid and dose-dependent release of glutamatefrom astrocytes was observed (Figs. 2A and 3). Gluta-mate release was observed immediately after NO ad-dition and was at least as fast as the response time ofthe method (a few seconds), but stopped after 10 –20 s,presumably because of the rapid decay of NO in suchsolutions (Bal-Price and Brown, 2001). In control ex-periments, in the absence of either L-glutamate dehy-drogenase, NADP, or astrocytes, there were no fluores-cence changes observed when nitric oxide was added. Glutamate release was mediated by nitric oxide be-cause NO scavenger PTIO (100 M) abolished it almostcompletely (Fig. 3). NO did not induce glutamate re-lease from cultured microglia (Fig. 3) using the sameconditions and the same number of cells, indicatingthat glutamate release is relatively specific to astro-cytes. Interestingly, NO-induced glutamate releasewas completely blocked by the preincubation of astro-cytes with either an extracellular Ca2ϩ chelator 4 mMEGTA (for 1 h; Figs. 2B and 3) or an intracellular Ca2ϩchelator 25 M BAPTA-AM (for 30 min; Figs. 2C and3). Since in the absence of calcium glutamate releasewas not observed, these results indicate that theNO-induced glutamate release from astrocytes wascalcium-dependent.
As there are studies suggesting that Ca2ϩ-dependent
glutamate release from astrocytes can occur by a pro-cess resembling neuronal exocytosis, we investigated
Fig. 2. Representative fluorescence traces of glutamate release
the effect of BoTx-C (1.5 nM), a specific toxin that
from rat cortical astrocytes in suspension induced by nitric oxide.
blocks the exocytotic release of neurotransmitters in
Without further additions (control), nitric oxide evoked rapid anddose-dependent release of glutamate from cultured astrocytes (21–28
neurons by cleavage of syntaxin (synaptic protein). It
DIV; A). Preincubation of astrocytes with either extracellular Ca2ϩ
has been shown that BoTx-C blocks bradykinin-in-
chelator 4 mM EGTA for 1 h (B) or an intracellular Ca2ϩ chelator 25
M BAPTA-AM for 30–40 min (C) or 1.5 nM botulinum toxin
duced calcium-dependent glutamate release from as-
(BoTx-C) for 24 h (D) blocked completely NO-induced glutamate re-
trocytes (Jeftinija et al., 1997). To determine whether
lease from astrocytes. Glutamate release induced by nitric oxide from
BoTx-C affects NO-induced glutamate release, astro-
astrocytes was not affected by preincubation with an inhibitor of the glutamate transporter 100 M t-PDC for 1 h (E). For each trace, ϳ 4 ϫ
cytes were incubated with BoTx-C (1.5 nM) for 16 or
106 astrocytes were added to 2 ml of Krebs-Hepes buffer in a fluorim-
24 h before NO addition as prolonged incubation with
eter cuvette followed by NADP and glutamate dehydrogenase (GD).
toxin is necessary to inhibit glutamate release from
Subsequent additions of 1 M NO, 2 M NO, 4 M NO, and 20 nmol
of glutamate were made (indicated by the four arrows).
astrocytes (Bezzi et al., 1998; Pasti et al., 2001). Thisprolonged incubation with BoTx-C (16 –24 h) did notcause any cell death as tested by Hoechst 33342 andpropidium iodide staining to assess whether any apo-
nificantly affected by preincubation of astrocytes with
ptotic or necrotic cells, respectively, were present. Pre-
an inhibitor of the glutamate transporter t-PDC (100
treatment of the astrocytes with BoTx-C for 16 h partly
M for 1 h; Figs. 2E and 3), suggesting that glutamate
decreased NO-induced glutamate release (data not
transporters are not required for NO-induced gluta-
shown), but 24-h incubation completely blocked it
mate release from cultured astrocytes. However, after
(Figs. 2D and 3). This suggests that syntaxin is re-
preincubation with t-PDC, NO-induced glutamate re-
quired for NO-evoked glutamate release and therefore
lease was not significantly dose-dependent. It is possi-
the release is probably due to exocytosis of vesicular
ble that the prolonged incubation with 100 M t-PDC
glutamate. NO-induced glutamate release was not sig-
(1 h) may perturb glutamate pools inside and outside
Fig. 3. Quantification of glutamate release evoked by 1, 2, or 4 M
icant glutamate release was seen in response to 1, 2, or 4 M NO.
NO from control astrocytes (nontreated; n ϭ 24) or preincubated with
Glutamate levels were measured using a continuous fluorometric
either 25 M BAPTA-AM (for 30 – 40 min; n ϭ 9), 4 mM EGTA (for 1 h;
assay. Results are expressed as the amount of glutamate released in
n ϭ 9), 100 M t-PDC (for 1 h; n ϭ 6), 1.5 nM botulinum toxin-C
nmol/106 cells means Ϯ SD. Levels that are statistically significantly
(BoTx-C; for 24 h; n ϭ 6), 10 M ODQ (for 20 min; n ϭ 6), 10 M
different from control astrocytes treated with 1, 2, or 4 M NO (first
indomethacin (INDO; for 20 min; n ϭ 6), or 100 M PTIO (n ϭ 6).
columns) are marked *P Ͻ 0.05, **P Ͻ 0.01, and ***P Ͻ 0.001.
Alternatively, the same number of microglia was added but no signif-
the cells, but it is clear that the inhibitor does not blockNO-induced glutamate release.
To provide further insights into the mechanism of
NO-induced glutamate release, astrocytes were alsopreincubated with 10 M ODQ (inhibitor of solubleguanylate cyclase) to test whether guanylate cyclase isinvolved. However, ODQ did not block NO-induced glu-tamate release from astrocytes, suggesting that guan-ylate cyclase is not involved (Fig. 3).
It has been reported that glutamate release from
astrocytes can be blocked by indomethacin, a cycloox-ygenase inhibitor (Bezzi and Volterra, 2001). However,in our studies, preincubation of astrocytes with indo-methacin (10 M) had no effect (Fig. 3), suggesting thatgeneration of prostaglandins is not involved in NO-
Fig. 4. Time course of 500 M DETA-NONOate–induced ATP re-
induced glutamate release from astrocytes. Indometh-
lease from ϳ 1.0 ϫ 106 of rat cortical astrocytes (21–28 DIV) prein-
acin, ODQ, or BoTx-C did not affect the glutamate
cubated with either PTIO (100 M; n ϭ 6), 25 M BAPTA-AM (n ϭ 6),
1.5 nM botulinum toxin-C (BoTx-C; n ϭ 6), or without additional
assays on their own and did not cause any cell death
treatment (n ϭ 9). ATP levels were determined luminometrically.
after the preincubation time as estimated by trypan
Results are expressed as concentration (nM in the medium) means Ϯ
SD. *P Ͻ 0.05, **P Ͻ 0.01, and ***P Ͻ 0.001 statistical significance of
difference from ATP release after the same time of DETA-NONOate
Release of glutamate was not observed after addition
treatment, but with no other additions (black bars).
of 2 mM azide (an inhibitor of cytochrome oxidase; datanot shown), suggesting again that NO-induced rapidrelease of glutamate from astrocytes was not mediated
Krebs-Hepes buffer (pH 7.4). Indeed, DETA-NONOate
caused a rapid increase of extracelluar ATP and after10 min was about 10-fold higher (5.8 Ϯ 0.7 nM) thanthe basal level (0.5 Ϯ 0.4 nM; Fig. 4). The ATP release
NO Induces Rapid Calcium-Dependent Release
was caused by nitric oxide since 100 M PTIO (NO
of Vesicular ATP From Astrocytes
scavenger) prevented it almost completely (Fig. 4).
Similarly to NO-mediated glutamate release, ATP
To test whether nitric oxide could also stimulate
release was also entirely calcium-dependent since
release of ATP, as it does in the case of glutamate,
30-min preincubation of cultured astrocytes with
cultured rat astrocytes (after shaking off microglia)
BAPTA-AM (25 M) completely blocked the ATP re-
were exposed to 500 M DETA-NONOate for 2.5, 5,
lease (Fig. 4). Since these results suggested that the
and 10 min and the level of ATP was measured in
vesicular mechanisms could be involved in NO-medi-
ated ATP release, the astrocytes were exposed to
by voltage-sensitive Ca2ϩ channels. To study further
BoTx-C (1.5 nM). Prolonged treatment with BoTx-C
the mechanisms of NO-induced [Ca2ϩ] increase, the
(24 –28 h) before exposure to DETA-NONOate (0.5
astrocytes were preincubated for 10 min with 10 M
mM) blocked NO-induced ATP release (Fig. 4). These
ODQ (guanylate cyclase inhibitor) before DETA-
results indicate that NO-induced ATP release from
NONOate application to determine whether NO-in-
cultured astrocytes was mediated by calcium-depen-
duced calcium entry into the cells could be mediated by
dent vesicular release, as in the case of NO-induced
the cGMP pathway. However, preincubation of astro-
glutamate release. Exposure of astrocytic culture to
cytes with ODQ did not have any influence on NO-
DETA-NONOate (500 M) for 2.5, 5.0, or 10 min,
induced increase of [Ca2ϩ] (Figs. 5D and 6), indicating
BoTx-C (1.5 nM) for 28 h, or BAPTA (25 M) for 30 min
that the NO-mediated Ca2ϩ entry is cGMP-indepen-
did not cause any cell death as determined by pro-
pidium iodide and Hoechst 33342 staining. Nitric Oxide Released From Nitric Oxide Induces a Rapid Biphasic Increase LPS/IFN-␥–Activated Astrocytes Causes High in Intracellular Calcium ([Ca2؉] ) Extracellular Glutamate Levels But Has Little Effect on Glutamate Uptake
To determine whether NO increased intracellular
calcium, astrocytes cultured on coverslips loaded with
Since exogenously added NO caused rapid glutamate
fura-2 were perfused with DETA-NONOate (500 M)
release, we tested whether endogenously produced NO
and intracellular calcium levels were monitored. The
from cytokine-activated astrocytes could elevate extra-
mean resting [Ca2ϩ] in astrocytes was 38.5 Ϯ 15.3 nM
(n ϭ 15; Fig. 6). Application of 500 M DETA-NONO-
Astrocytes (21–28 DIV, confluent) were exposed for
ate resulted in a rapid, transient (1–3 min) increase in
18 h to LPS and IFN-␥, which leads to high levels of NO
[Ca2ϩ] (to 141.2 Ϯ 17.8 nM; Figs. 5 and 6) followed by
production from iNOS (Bal-Price and Price, 2001) in
return to a lower, but still elevated, level of calcium
the presence or absence of an iNOS inhibitor 1400W
(63.1 Ϯ 8.3 nM) sustained for at least 10 min (Figs. 5
(25 M). After 18 h of activation, glutamate levels in
and 6). The observed increase in [Ca2ϩ] in astrocytes
the medium were measured using a colorimetric
was indeed induced by NO and was completely and
method and compared with nonactivated astrocytes. In
rapidly reversible; after addition of 100 M PTIO (NO
the case of activated astrocytes, high levels of gluta-
scavenger), the level of [Ca2ϩ] dropped to the level
mate were present in the medium (7.38 Ϯ 1.6 M) after
observed before DETA-NONOate application (Figs. 5A
18 h of activation as compared with very low level of
glutamate in the medium of control astrocytes (0.43 Ϯ
To test whether the nitric oxide-induced [Ca2ϩ] rise
0.06 M; Fig. 7). In the presence of the iNOS inhibitor
resulted from calcium influx across the plasma mem-
(25 M 1400W), glutamate was still present in the
brane or mobilization from intracellular stores, calcium
medium but at a lower level (3.72 Ϯ 0.6 M). These
was removed from Krebs-Hepes buffer prior to DETA-
results are compatible with NO from activated astro-
NONOate application. In this case, only a transient
cytes inducing glutamate release. However, the same
increase in [Ca2ϩ] was observed after DETA-NONOate
result could be caused by a decreased capacity of acti-
application (Fig. 5E), suggesting that extracellular cal-
vated astrocytes for glutamate uptake.
cium was required for the generation and maintenance
To determine whether nitric oxide produced by the
of the sustained [Ca2ϩ] plateau observed during con-
proinflammatory cytokine-activated astrocytes could
tinued presence of extracellular calcium. The specific
attenuate astrocytic capacity for glutamate uptake,
inhibitor of capacitative calcium entry (CCE) 1 M
100 M exogenous glutamate from stock solution was
gadolinum (Moneer and Taylor, 2002) immediately
added to the astrocytes after 18 h of activation in the
blocked the increase in [Ca2ϩ] observed after DETA-
presence and absence of iNOS inhibitor (25 M
NONOate application (Fig. 5B). These results suggest
1400W), and then samples of the medium were taken
that the biphasic [Ca2ϩ] increase induced by nitric
at various intervals of time (1, 10, 30 min or 1, 4, and
oxide could be separated into two components: an ini-
24 h) and glutamate content was measured. In the
tial transient component that is due to calcium mobi-
control cultures, glutamate concentrations showed a
lization from intracellular stores and a plateau compo-
rapid initial decline; within 30 min glutamate concen-
trations dropped to 6.7 Ϯ 1.46 M (Fig. 7) and after 1 h
Since several studies have described the expression
reached values below 1 M. These data show that
of voltage-activated calcium channels in astrocytes, we
control astrocytes were capable of taking up glutamate
tested whether nitric oxide-induced Ca2ϩ entry could
rapidly and efficiently. LPS/IFN-␥–activated astro-
be partially mediated by voltage-gated Ca2ϩ channels.
cytes removed glutamate from the medium at a similar
However, 10 M verapamil (blocker of L-type Ca2ϩ
rate to nonactivated (or 1400W-treated) astrocytes.
channels) did not affect the [Ca2ϩ] plateau induced by
However, the level of glutamate in the medium of ac-
DETA-NONOate (Figs. 5C and 6), suggesting that NO-
tivated astrocytes was always higher than in the con-
induced Ca2ϩ entry into astrocytes was not mediated
trol culture at each time point both before and after
glutamate addition (Fig. 7). After 24 h, glutamate was
a steady-state level lower than in the absence of 1400W
still present in the medium of activated astrocytes
(but higher than in nonactivated astrocytes).
(7.8 Ϯ 1.5 M) at concentration similar to that prior the
These results suggest that the elevated extracellular
addition of 100 M glutamate. In the presence of the
glutamate level maintained by activated astrocytes is
iNOS inhibitor (1400W), glutamate uptake was again
due to increased glutamate release, with little or no
similar, but after 24 h the glutamate level returned to
DISCUSSION
It has previously been shown that NO can cause
acute glutamate release from neurons or synaptosoms(Sequeira et al., 1997; McNaught and Brown, 1998;Bal-Price and Brown, 2001), and we have attributedthis to NO inhibition of mitochondrial respiration caus-ing a fall in ATP, inhibition of the sodium pump, andreversal of the glutamate transporter. Evidence for thismechanism was that the time, oxygen, and NO depen-dence of glutamate release and respiratory inhibitionwere similar, glutamate release was calcium-indepen-dent, and specific respiratory inhibitors caused a sim-ilar fall in ATP and glutamate release (McNaught andBrown, 1998; Bal-Price and Brown, 2001). Sequeira etal. (1997) found a similar calcium-independent gluta-mate release from synaptosomes induced by NO, asso-ciated with a decrease in ATP/ADP ratio and inhibitedby t-PDC (an inhibitor of the glutamate transporter). In contrast, we find here that NO-induced glutamaterelease from astrocytes is completely calcium-depen-dent and is not replicated by respiratory inhibitors. The reason NO does not cause energy depletion-in-duced glutamate release from astrocytes is probablybecause although it inhibits astrocytic respiration(Brown et al., 1995), it does not cause a fall in astrocyticATP (Bal-Price and Brown, 2001), presumably becauseof the relatively high glycolytic capacity of astrocytes(Pauwels et al., 1985; Peuchen et al., 1997). Our findingthat t-PDC did not have any significant effect on NO-induced glutamate release from astrocytes confirmsthat this release is not mediated by reversal of theglutamate transporter.
In the case of neurons, it has been proposed that NO
causes glutamate release by directly modifying the exo-cytotic machinery of synaptic vesicles. This was basedon the finding that neuronal glutamate release inducedby NO was calcium-independent but inhibited by butu-
Fig. 5. Representative traces of NO-induced increase in intracellu-
lar calcium [Ca2ϩ] in astrocytes after perfusion with 500 M DETA-
NONOate. The increase was caused by NO released from DETA-NONOate because 100 M PTIO (NO scavenger) completely reversed
the increase (A). The sustained component of Ca2ϩ entry was also inhibited by the inhibitor of capacitative calcium entry, 1 M gado-
linium (Gd3ϩ; B), but was unaffected by the L-type Ca2ϩ channel inhibitor, 10 M verapamil (C) or by the soluble guanylate cyclase
inhibitor, 10 M ODQ (D). The sustained level of [Ca2ϩ] induced by
DETA-NONOate (500 M) was abolished by removal of extracellular
Ca2ϩ, whereas the initial mobilization was unaffected (E). The mea- surements of [Ca2ϩ] were performed in fura-2–loaded astrocytes us-
ing one coverslip (ϳ 0.15 ϫ 106 cells). Bars represent the time of
perfusion with the indicated substances. DETA-NONOate (500 M)
was used in all the above experiments.
nisms are not relevant in astrocytes, at least in ourconditions.
Astrocytes have a glutamate release mechanism sim-
ilar to that of neurons, mediated by exocytosis of vesi-cles loaded with glutamate (Araque et al., 2000; Bezziand Volterra, 2001). This exocytosis is triggered byincreased cytosolic calcium (often in the form of cal-cium waves) (Bezzi et al., 1998; Araque et al., 2000)and is inhibited by botulinum neurotoxin C or B (Jeft-inja et al., 1997; Araque et al., 2000). As we found thatNO-induced glutamate and ATP release from astro-cytes
BoTx-C, it seems likely that this release is mediated byvesicular exocytosis.
It has previously been shown that NO induces acute
increases of cytosolic calcium (and calcium waves) inastrocytes, possibly via activation of soluble guanylate
Fig. 6. Quantification of the increase in intracellular calcium in-
cyclase (Willmott et al., 2000a). In our present studies,
duced by DETA-NONOate (500 M) in cultured rat astrocytes in the
absence (n ϭ 24) or presence of 100 M PTIO (n ϭ 3), 10 M ODQ (n ϭ
we show that the application of DETA-NONOate to
6), 10 M verapamil (n ϭ 3), 1 M gadolinium (Gd3ϩ) (n ϭ 3), or in the
fura-2–loaded astrocytes caused a very rapid increase
absence of extracellular Ca2ϩ (n ϭ 3). Values represent means Ϯ SD.
of [Ca2ϩ] , but this was not blocked by ODQ (a soluble
**P Ͻ 0.01 from calcium concentration in astrocytes exposed to
DETA-NONOate (500 M) alone for 11 min in Krebs-Hepes buffer
guanylate cyclase inhibitor), suggesting that soluble
guanylate cyclase was not involved. The discrepancybetween our studies and the results described by Will-mot et al. (2000a) is possibly due to the fact that intheir studies LY83583 was used to block guanylatecyclase activity, but this compound is known also toinactivate nitric oxide (Barbier and Lefebvre, 1992), incontrast to ODQ, which does not affect NO levels. cGMP-independent mechanisms of NO-induced in-crease in [Ca2ϩ] were also shown in C6 glioma (Bow-
man et al., 2001) and Bergmann glial cells (Matyash etal., 2001).
Since gadolinum returned the [Ca2ϩ] from the pla-
teau level to the basal level, these results suggest thatCa2ϩ entry occurs through CCE mechanism in astro-cytes. However, gadolinum can also inhibit L- and N-type voltage-dependent calcium channels that areexpressed on some astrocytes (Sontheimer, 1994;Agrawal et al., 2000). Ca2ϩ entry across the plasmamembrane was not mediated by L-type voltage-sensi-tive Ca2ϩ channels because verapamil did not affect
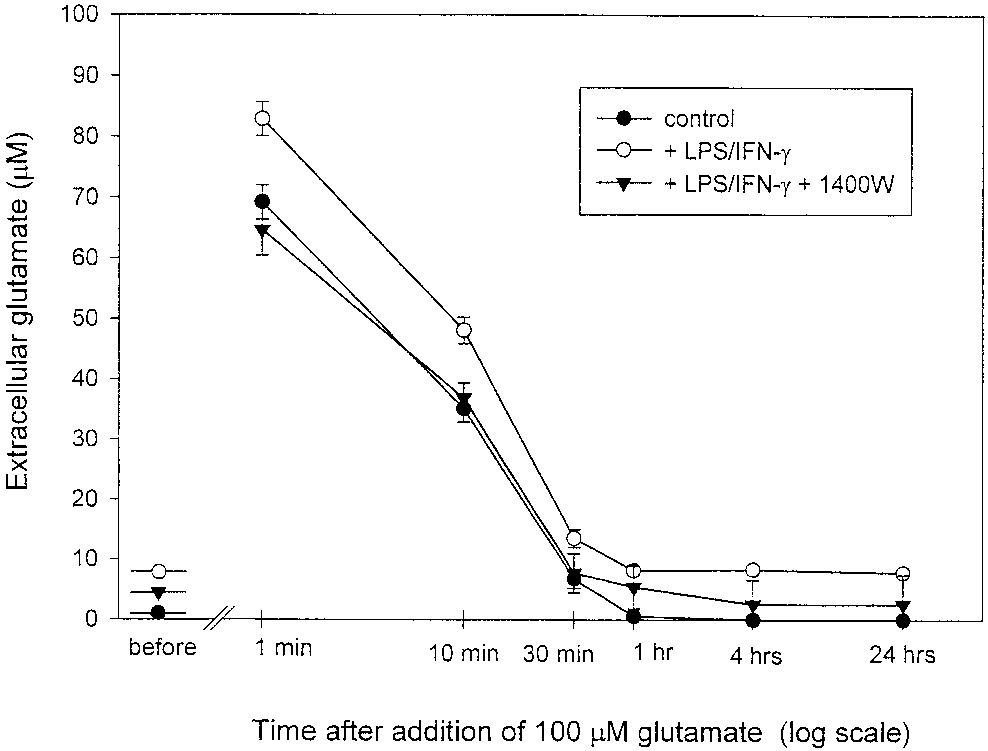
Fig. 7. Extracellular glutamate levels before and after addition of
[Ca2ϩ] levels but we cannot rule out that N-type chan-
100 M glutamate to the medium of nonactivated (control) and LPS/
IFN-␥–activated astrocytes in the presence or absence of iNOS inhib-
nels contribute to calcium entry. As in other nonexcit-
itor 1400W (25 M; n ϭ 3). Samples of media were taken at the points
able cells exposed to agents mobilizing calcium from
indicated and the glutamate content was determined. Note that the
endoplasmic reticulum (Matsumoto et al., 1986; Kot-
levels of glutamate in the medium of astrocytes prior to glutamateadditions were 0.43 Ϯ 0.06 M (control, nonactivated), 7.38 Ϯ 1.6 M
likoff et al., 1987), the initial component of the biphasic
(activated in the absence of 1400W; P Ͻ 0.001 from control), and
[Ca2ϩ] increase observed after application of DETA-
3.72 Ϯ 0.6 M (activated in the presence of 1400W; P Ͻ 0.01 from
NONOate was insensitive to the removal of extracellu-
activated without 1400W). Twenty-four hours after glutamate addi-tion (100 M), the levels of glutamate were 0.0 M in control culture,
lar Ca2ϩ, indicating that it was due to mobilization of
7.8 Ϯ 1.5 M in the medium of activated astrocytes without 1400W
Ca2ϩ from intracellular stores. Depletion of intracellu-
(P Ͻ 0.001 from control culture), and 2.58 Ϯ 0.8 M in activated
astrocyte with 1400W (P Ͻ 0.05 from activated astrocytes without
lar Ca2ϩ could provide a sufficient signal for activation
1400W). Values represent means Ϯ SD.
of Ca2ϩ entry through the plasma membrane as extra-cellular Ca2ϩ is required for the generation and main-tenance of the sustained [Ca2ϩ] plateau during pro-
linum neurotoxins A, C, and F and associated with
longed application of DETA-NONOate. The NO-
covalent modifications of vesicle proteins (Meffert et
induced glutamate release from astrocytes found here
al., 1996). However, our finding that NO-induced glu-
was fast—faster than the response time of the contin-
tamate (and ATP) release in astrocytes is completely
uous assay (Fig. 2)—and terminated when the added
calcium-dependent suggests that such direct mecha-
NO was likely to have disappeared from the medium,
so further NO addition caused more glutamate release.
mate release but we have not studied whether the
This suggests that the effect was mediated by NO it-
NO-induced ATP release potentiates glutamate release
self, rather than one of its derivatives (peroxynitrite,
or vice versa, as has been suggested in other studies
NO , N O , or S-nitrosothiols), as it would take time for
(Queiroz et al., 1999). However, the NO-induced in-
these derivatives to be produced from NO. Virtually all
crease in extracellular ATP observed in our studies is
rapid, reversible effects of NO are mediated by NO
rather low (5.8 Ϯ 0.7 nM), and whether it could play a
binding to hemeproteins, such as soluble guanylate
significant role in intercellular signaling needs further
cyclase or cytochrome oxidase (Xu et al., 1998; Eu et al.,
2000). However, neither of these hemeproteins appears
Physiologically, in the brain NO is mainly produced
to mediate the NO-induced glutamate release from as-
by nNOS in subpopulation of neurons, and this nNOS
trocytes as release was not prevented by an inhibitor of
is transiently activated by calcium elevations during
soluble guanylate cyclase (ODQ) and release was not
neuronal activity (Vincent, 1994; Baader and Schilling,
induced by an inhibitor of cytochrome oxidase (azide).
1996; Baltrons et al., 1997). This suggests the possibil-
Willmott et al. (2000a) found that NO-induced calcium
ity that NO released from active neurons may cause
waves in astrocytes were eliminated by high concen-
calcium waves in and glutamate (and ATP) release
trations of ryanodine, suggesting that NO activates
from surrounding astrocytes, and thus might be in-
ryanodine receptor-mediated calcium release from en-
volved in neuronal-astrocytic communication and could
doplasmic reticulum. NO can elevate calcium in heart
particularly affect synaptic transmission as astrocytes
and skeletal muscle by S-nitrosating or oxidizing ryan-
and their processes surround synapses (Parpura et al.,
odine receptors (Eu et al., 2000). This has not been
1994; Bezi and Volterra, 2001). eNOS is expressed in
reported in astrocytes, but as astrocytes express the R3
some astrocytes and has been suggested to be involved
isoform of ryanodine receptors (Matyash et. al, 2002), a
in initiation and propagation of calcium waves (Will-
possible mechanism by which NO elevates calcium in
mott et al., 2000b), and thus potentially astrocytic
astrocytes is by S-nitrosating or promoting thiol oxida-
eNOS might regulate glutamate and ATP release from
tion of the ryanodine receptor to cause release from the
astrocytes. Whether neurons or astrocytes locally re-
lease sufficient NO to cause significant glutamate or
The mechanisms by which ATP is released from as-
ATP release from astrocytes is unclear, but levels of
trocytes are still unclear. In our present studies, NO-
NO up to 100 nM have been measured by electrode in
induced ATP release was entirely calcium-dependent
brain slices induced by electrical stimulation (Shibuki
and blocked by BoTx-C, suggesting vesicular exocyto-
and Kimura, 1997), up to 1 M NO released from
sis. Indeed, ATP has been identified as a storage com-
cultured aortic endothelial cells induced by bradykinin
ponent of astrocytic vesicles released from cultured
(Clementi et al., 1999), and up to 1– 4 M has been
astrocytes (Maienschein et al., 1999) by receptor-medi-
measured in rat brain in vivo during ischemia and
ated mechanisms (Queiroz et al., 1997). Calcium-de-
reperfusion (Malinski et al., 1993). The NO released
pendent ATP release from astrocytes was also reported
during ischemia or reperfusion might trigger extensive
in response to excitatory amino acids like N-methyl-D-
glutamate release from astrocytes, which could contrib-
aspartate (NMDA) and kainite (Queiroz et al., 1999).
ute to the known pathological glutamate release in
However, other mechanisms of ATP release have been
these conditions (Szatkowski and Attwell, 1994; Rossi
reported, for example, through connection hemi-
channels (Cotrina et al., 1998b; Stout et al., 2002) or
In a wide range of brain pathologies, astrocytes and
calcium-independent mechanism after stimulation of
microglia become activated by inflammatory mediators
alpha-amino-3-hydroxy-5-methylisoxazole-4-propio
to express iNOS (Wisniewski et al., 1998; Murphy,
(ATP) receptors (Queiroz et al., 1999).
2000). In culture, activated glia can kill cocultured
Recently, it has been shown that ATP released from
neurons by NO and excitotoxic mechanisms (Chao,
astrocytes mediates increases in cytosolic calcium and
1996; Kingham et al., 1999; Bal-Price and Brown,
propagation of calcium waves in glia (Cotrina et al.,
2001). Such activated astrocytes and microglia contin-
1998a; James and Butt, 2001). ATP-dependent calcium
uously produce an extracellular NO level of about 1 M
signaling in astrocytes is mediated by metabotropic
(Brown et al., 1995; Bal-Price and Brown, 2001), suffi-
purinergic P2Y receptors (coupled to calcium release
cient to cause substantial glutamate release from as-
from internal stores) (Guthrie et al., 1999; Fam et al.,
trocytes according to our results here. And indeed we
2000). ATP apparently also mediates calcium signaling
have shown here that activated astrocytes maintain a
between astrocytes and microglia through P2X puri-
substantially higher extracellular glutamate level,
nergic receptor located on microglia (Verderio and Mat-
which appears to be due to enhanced glutamate release
teoli, 2001). ATP-mediated increase in intracellular
rather than reduced uptake. It has previously been
calcium was also observed after the mechanical stimu-
reported (Ye and Sontheimer, 1998) that uptake of
lation of astrocytes in culture (Stout et al., 2002). In-
glutamate into astrocytes is inhibited by NO or inflam-
terestingly, ATP can also cause a dose-dependent re-
matory activation; however, the level of inhibition re-
lease of glutamate and aspartate from cultured rat
ported was relatively small (about 30%), such that it
astrocytes (Jeremic et al., 2001). In the present studies,
would be unlikely to contribute substantially to an
we have shown that NO induced both ATP and gluta-
elevated extracellular glutamate level. We found no
obvious inhibition of glutamate uptake by activated
Dinerman JL, Dawson TM, Schell MJ, Snowman A, Snyder SH. 1994.
astrocytes, but our data are not sufficient to rule out a
Endothelial nitric oxide synthase localized to hippocampal pyrami-dal cells: implications for synaptic plasticity. Proc Natl Acad Sci
small inhibition of the order of 30%. According to other
data (Patneau and Mayer, 1990), the level of extracel-
Eddleston M, Mucke L. 1993. Molecular profile of reactive astrocytes:
implications for their role in neurologic disease. Neuroscience 54:
lular glutamate we find maintained by activated astro-
cytes would be sufficient to activate NMDA receptors
Eu IP, Sun I, Xu L, Stamler JS, Meissner G. 2000. The skeletal muscle
and therefore might contribute to the mechanisms by
calcium release channel: coupled O sensor and NO signaling func-
which activated glia kill neurons in coculture. Thus,
Fam SR, Gallagher CJ, Salter MW. 2000. P2Y1, purinoreceptor-me-
NO-induced release of glutamate from astrocytes po-
diated Ca2ϩ signaling and Ca2ϩ waves propagation in dorsal spinal
tentially might contribute to neuronal death during all
cord astrocytes. J Neurosci 20:2800 –2808.
Garthwaite J, Boulton CL. 1995. Nitric oxide signaling in the central
the inflammatory, infectious, ischemic, and neurode-
nervous system. Ann Re Physiol 57:683–706.
generative diseases where iNOS has been shown to
Goodwin Jl, Uemura E, Cunnick JE. 1995. Microglial release of nitric
be expressed in glia (Eddleston and Mucke, 1993;
oxide by the synergistic action of beta-amyloid and IFN-gamma. Brain Res 692:207–214.
Guthrie PB, Knappenberger J, Segal M, Bennett MVL, Charles AC.
1999. ATP released from astrocytes mediates glial calcium waves. J Neurosci 19:520 –528.
Heales SJR, Bolanos JP, Stewart VC, Brookes PS, Land JM, Clark
ACKNOWLEDGMENT
JB. 1999. Nitric oxide, mitochondria and neurological disease. Bio-chim Biophys Acta 1410:215–228.
Hewett SJ, Csernansky CA, Choi DW. 1994. Selective potentiation of
The authors thank Dr Edward Bamtpon for prepar-
NMDA-induced neuronal injury following induction of astrocytic
Ignarro LJ. 2000. Nitric oxide: biology and pathology. San Diego, CA:
Innocenti B, Parpura V, Haydon PG. 2000. Imaging extracellular
REFERENCES
waves of glutamate during calcium signalling in cultured astro-cytes. J Neurosci 20:1800 –1808.
James G, Butt AM. 2001. P2X and P2Y purinoreceptors mediate
Agrawal SK, Nashmi R, Fehlings MG. 2000. Role of L- and N-type
ATP-evoked calcium signalling in optic nerve glia in situ. Cell
calcium channels in the pathophysiology of traumatic spinal cord
white matter injury. Neuroscience 99:179 –188.
Jeftinja SD, Jeftinija KV, Stefanovic G. 1997. Cultured astrocytes
Araque A, Li N, Doyle RT, Haydon PG. 2000. SNARE protein-depen-
express proteins involved in vescicular glutamate release. Brain
dent glutamate release from astrocytes. J Neurosci 20:666 – 673.
Baader SL, Schilling K. 1996. Glutamate receptors mediate dynamic
Jeremic A, Jeftinija K, Stevanovic J, Glavaski A, Jeftinija S. 2001.
regulation of nitric oxide synthase expression in cerebellar granule
ATP stimulates calcium-dependent glutamate release from cul-
tured astrocytes. J Neurochem 77:664 – 675.
Bal A, Bachelot T, Savasta M, Manier M, Verna JM, Benabid AL,
Kingham PJ, Cuzner ML, Pocock JM. 1999. Apoptotic pathways mo-
Feuerstein C. 1994. Evidence for dopamine D
bilized in microglia and neurons as a consequence of chromogranin
expression by striatal astrocytes in culture: in situ hybridization
A-induced microglial activation. J Neurochem 73:538 –547.
and polymerise chain reaction studies. Mol Brain Res 23:204 –212.
Knott C, Stern G, Wilkin GP. 2000. Inflammatory regulators in Par-
Bal-Price A, Brown GC. 2001. Inflammatory neurodegeneration me-
kinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2.
diated by nitric oxide from activated glia-inhibiting neuronal respi-
ration, causing glutamate release and excitotoxicity. J Neurosci
Kotlikoff ML, Murray RK, Reynolds EE. 1987. Histamine-induced
calcium release and phorbol antagonism in cultured airway smooth
Baltrons MA, Saadoun S, Agullo L, Garcia A. 1997. Regulation by
muscle cells. Am J Physiol 253:561–566.
calcium of the nitric oxide/cyclic GMP system in cerebellar granule
Kreutzberg GW. 1996. Microglia: a sensor for pathological events in
cells and astroglia in culture. J Neurosci Res 49:333–341.
Barbier AJ, Lefebvre RA. 1992. Effect of LY 83583 on relaxation
Lee SC, Zhao ML, Hirano A, Dickson DW. 1999. Inducible nitric oxide
induced by non-adrenergic non-cholinergic nerve stimulation and
synthase immunoreactivity in the Alzheimer disease hippocampus:
exogenous nitric oxide in the rat gastric fundus. Eur J Pharmacol
association with Hirano bodies, neurofibrillary tangles, and senile
plaques. J Neuropathol Exp Neurol 58:1163–1169.
Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan
Leist M, Fava E, Montecucco C, Nicotera P. 1997. Peroxynitrite and
T, Volterra A. 1998. Prostaglandins stimulate calcium-dependent
nitric oxide donors induce neuronal apoptosis by eliciting autocrine
glutamate release in astrocytes. Nature 391:281–285.
excitotoxicity. Eur J Neurosci 9:1488 –1498.
Bezzi P, Volterra A. 2001. A neuron-glia signalling network in the
Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O,
active brain. Curr Opin Neurobiol 11:387–394.
Ashe KH, Frautschy SA, Cole GM. 2000. Ibuprofen suppresses
Bolanos JP, Almeida A. 1999. Role of nitric oxide in brain hypoxia-
plaque pathology and inflammation in a mouse model for Alzhei-
ischaemia. Biochim Biophys 1411:415– 436.
mer’s disease. J Neurosci 20:5709 –5714.
Bowman CL, Yohe L, Lohr JW. 2001. Regulation of cytoplasmic cal-
Loihl AK, Murphy S. 1998. Expression of NOS-2 in glia associated
cium levels by two nitric oxide receptors. Am J Physiol 281:876 –
with CNS pathology. Prog Brain Res 118:253–267.
Maienschein V, Marxen M, Volknandt W, Zimmermann H. 1999. A
Brown GC, Bolanos JP, Heals SJ, Clark JB. 1995. Nitric-oxide pro-
plethora of presynaptic proteins associated with ATP-storing or-
duced by activated astrocytes rapidly and reversibly inhibits cellu-
ganelles in cultured astrocytes. Glia 26:233–244.
lar respiration. Neurosci Lett 193:201–204.
Malinski T, Bailey F, Zhang ZG, Chopp M. 1993. Nitric oxide mea-
Chao CC. 1996. Cytokine-stimulated astrocytes damage human neu-
sured by a porphyrinic microsensor in rat brain after transient
rons via a NO mechanism. Glia 16:276 –284.
middle cerebral artery occlusion. J Cereb Blood Flow Metab 13:355–
Clementi E, Brown GC, Foxwell N, Moncada S. 1999. On the mecha-
nism by which vascular endothelial cells regulate their oxygen
Matsumoto T, Kanaide H, Nishimura , Shogakiuchi Y, Kobayashi S,
consumption. Proc Natl Acad Sci USA 96:1559 –1562.
Nakamura. 1986. Histamine activates H receptors to induce cyto-
Cohen J, Wilkin GP. 1995. Neuronal cell culture. New York: Oxford
solic free calcium transients in cultured vascular smooth muscle
cells from rat aorta. Biochem Biophysic Res 135:172–177.
Cotrina ML, Lin JHC, Nedergaard M. 1998a. Cytoskeletal assembly
Matyash V, Filippov V, Mohrhagen K, Kettenmann H. 2001. Nitric
and ATP release regulate astrocytic calcium signaling. J Neurosci
oxide signals parallel fiber activity to Bergmann glial cells in the
mouse cerebellar slice. Mol Cell Neurosci 18:664 – 670.
Cotrina ML, Lin JHC, Nedergaard M. 1998b. Connexins regulate
Matyash M, Matyash V, Nolte C, Sorrentino V, Kettenmann H. 2002.
calcium signaling by controlling ATP release. Proc Natl Acad Sci
Requirement of functional ryanodine receptor type 3 for astrocytes
Mazzanti M, Sul JY, Haydon PG. 2001. Glutamate on demand; astro-
Rossi DJ, Oshima T, Attwell D. 2000. Glutamate release in severe
cytes as a ready source. Neuroscientist 7:396 – 405.
brain ischaemia is mainly by reversed uptake. Nature 403:316 –321.
McNaught KSP, Brown GC. 1998. Nitric oxide causes glutamate
Scemes E. 2000. Components of astrocytic intercellular calcium sig-
release from brain synaptosomes following inhibition of mitochon-
drial function. J Neurochem 70:1541–1546.
Sequeira S, Ambrosio A, Malva JO, Carvalho AP, Carvalho CM. 1997.
McGeer EG, McGeer PL. 1995. Brain inflammation in Alzheimer
Modulation of glutamate release from rat hippocampal synapto-
disease and the therapeutic implications. Curr Pharm Des 10:821–
somes by nitric oxide. Nitric Oxide Biol Chem 1:315–329.
Shibuki K, Kimura S. 1997. Dynamic properties of nitric oxide release
McGeer PL, Schulzer M, McGeer EG. 1996. Arthritis and anti-inflam-
from parallel fibers in rat cerebellar slices. J Physiol 15:443– 452.
matory agents as possible protective factors for Alzheimer’s disease:
Sontheimer H. 1994. Voltage-dependent ion channels in glial cells.
a review of 17 epidemiologic studies. Neurology 47:425– 432.
Meffert MK, Premack BA, Schulman H. 1994. Nitric oxide stimulates
Stout CE, Constantin JL, Naus CC, Charles AC. 2002. Intercellular
Caϩ2-independent synaptic vesicle release. Neuron 12:1235–1244.
calcium signaling in astrocytes via ATP release through connexin
Meffert MK, Calakos NC, Scheller RH, Schulman H. 1996. Nitric
hemichannels. J Biol Chem 10:1074 –1084.
oxide modulates synaptic vesicle docking/fusion reactions. Neuron
Szatkowski M, Attwell D. 1994. Triggering and execution of neuronal
death in brain ischaemia: two phases of glutamate release by dif-
Moneer Z, Taylor CW. 2002 Reciprocal regulation of capacitative and
ferent mechanisms. TINS 17:359 –365.
non-capacitative Ca2ϩ entry in A7r5 vascular smooth muscle cells:
Taupenot L, Ciesielski-Treska J, Urlich G, Chasserot-Golaz S, Aunis
only the latter operates during receptor activation. Biochem J 362:
D, Bader MF. 1996. Chromogranin A triggers a phenotypic trans-
formation and the generation of nitric oxide in brain microglial
Murphy S. 2000. Production of nitric oxide by glial cells: regulation
Trabace L, Kendrick KM. 2000. Nitric oxide can differentially modu-
and potential roles in the CNS. Glia 29:1–14.
late striatal neurotransmitter concentrations via soluble guanylate
Nicholls DG, Sihra TS, Sanchez-Prieto J. 1987. Calcium-dependent
cyclase and peroxynitrite formation. J Neurochem 75:1664 –1674.
and -independent release of glutamate from synaptosomes moni-
Wa J, Food MR, Gabathuler R, Rothenberger S, Yamada T, Yasuhara
tored by continuous fluorometry. J Neurochem 49:50 –57.
O, McGeer PL. 1996. Reactive microglia specifically associated with
Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG.
amyloid plaques in Alzheimer’s disease brain tissue express mela-
1994. Glutamate-mediated astrocyte-neuron signalling. Nature
notransferrin. Brain Res 712:122–126.
Wallace MN, Geddes JG, Farquhar DA, Masson MR. 1997. Nitric
Pasti L, Zonta M, Pozzan T, Vicini S, Carmingnoto G. 2001. Cytosolic
oxide synthase in reactive astrocytes adjacent to beta-amyloid
calcium oscillations in astrocytes may regulate exocytotic release of
glutamate. J Neurosci 21:477– 484.
Verderio C, Matteoli M. 2001. ATP mediates calcium signaling be-
Patneau DK, Mayer ML. 1990. Structure-activity relationships for
tween astrocytes and microglial cells: modulation by IFN-gamma.
aminoacid transmitter candidates acting at N-methyl-D-aspartate
and quisqualate receptors. J Neurosci 10:2385–2399.
Vincent SR. 1994. Nitric oxide: a radical neurotransmitter in the
Pauwels PJ, Opperdoes FR, Trouet A. 1985. Effects of antimycin,
central nervous system. Prog Neurobiol 42:129 –160.
glucose deprivation, and serum on cultures of neurons, astrocytes,
Willmott NJ, Wong K, Strong A. 2000a. A fundamental role for the
and neuroblastoma cells. J Neurochem 44:143–148.
nitric-oxide-G-kinase signaling pathway in mediating intracellular
Peuchen S, Bolanos JP, Heals SJR, Almeida A, Duchen MR, Clark JB.
Ca2ϩ waves in glia. J Neurosci 20:1767–1779.
1997. Interrelations between astrocytes function, oxidative stress
Willmott NJ, Wong K, Strong AJ. 2000b. Intercellular Ca2ϩ waves in
and antioxidant status within the central nervous system. Prog
rat hippocampal slice and dissociated glial-neuron cultures medi-
ated by nitric oxide. FEBS Lett 487:239 –247.
Prast H, Philippu A. 2001. Nitric oxide as modulator of neuronal
Wisniewski HM, Wegiel J, Wisniewski T. 1998. Pathogenesis of amy-
function. Prog Neurobiol 64:51– 68.
loid- plaques: activated microglia the cause of fibrillar amyloid
Queiroz G, Gebicke-Haerter PJ, Schobert A, Starke K, von Kugelgen
formation and neuropil degeneration. Neurosci News 1:30 –34.
I. 1997. Release of ATP from cultured rat astrocytes elicited by
Xu L, Eu JP, Meissner G, Stamler JS. 1998. Activation of the cardiac
glutamate receptor activation. Neuroscience 78:1203–1208.
calcium release channel, rayanodine receptor, by poly-S-nitrosyla-
Queiroz G, Meyer DK, Meyer A, Starke K, von Kugelgen I. 1999. A
study of the mechanism of the release of ATP from rat cortical
Ye Z, Sontheimer H. 1998. Glial glutamate transport as target for
astroglial cells evoked by activation of glutamate receptors. Neuro-
nitric oxide: consequences for neurotoxicity. Prog Brain Res 118:
Proza [i prozacul DEZINHIBAREA, lejeritatea, F`r` raport cu preocup`rile anteri- nele, cu trecerile lor nenum`rate de-o le în coam`, trag tot mai tare, m` smu-oare, formînd partea cea mai întins` a cesc, m` scutur, ]op`i ca o descreierat`. Prozacului , tabletele din Suplimentul de cit, ce pare pierdut în ocoli[uri [i opriri cultur` sînt scrise de un autor ce se ia de plîns.
Ausgabe: Januar 2006 zuletzt geändert und ergänzt: März 2007 Technische Regeln Arbeitsplatzgrenzwerte Gefahrstoffe Die Technischen Regeln für Gefahrstoffe (TRGS) geben den Stand der Technik,Arbeitsmedizin und Arbeitshygiene sowie sonstige gesicherte wissenschaftlicheErkenntnisse für Tätigkeiten mit Gefahrstoffen, einschließlich deren Einstufung undKennzeichnung, wieder. Sie w
 proximately 2– 4 min later, glutamate release was ini-tiated by addition of 1, 2, or 4 l of NO-saturated waterat 20°C (2 mM). NO-saturated water was prepared bybubbling oxygen-free nitrogen through distilled waterin a glass vial with a rubber seal, then bubbling withNO gas until the water was NO-saturated. Glutamaterelease was calibrated by adding a known amount ofglutamate at the end of each assay. The relationshipbetween glutamate concentration and NADPH fluores-cence was tested by adding 5, 10, or 15 M of glutamateto the assay and was found to be roughly linear overthis range. The amplitude of the NADPH fluorescencereached a plateau after 10 –20 s.
proximately 2– 4 min later, glutamate release was ini-tiated by addition of 1, 2, or 4 l of NO-saturated waterat 20°C (2 mM). NO-saturated water was prepared bybubbling oxygen-free nitrogen through distilled waterin a glass vial with a rubber seal, then bubbling withNO gas until the water was NO-saturated. Glutamaterelease was calibrated by adding a known amount ofglutamate at the end of each assay. The relationshipbetween glutamate concentration and NADPH fluores-cence was tested by adding 5, 10, or 15 M of glutamateto the assay and was found to be roughly linear overthis range. The amplitude of the NADPH fluorescencereached a plateau after 10 –20 s.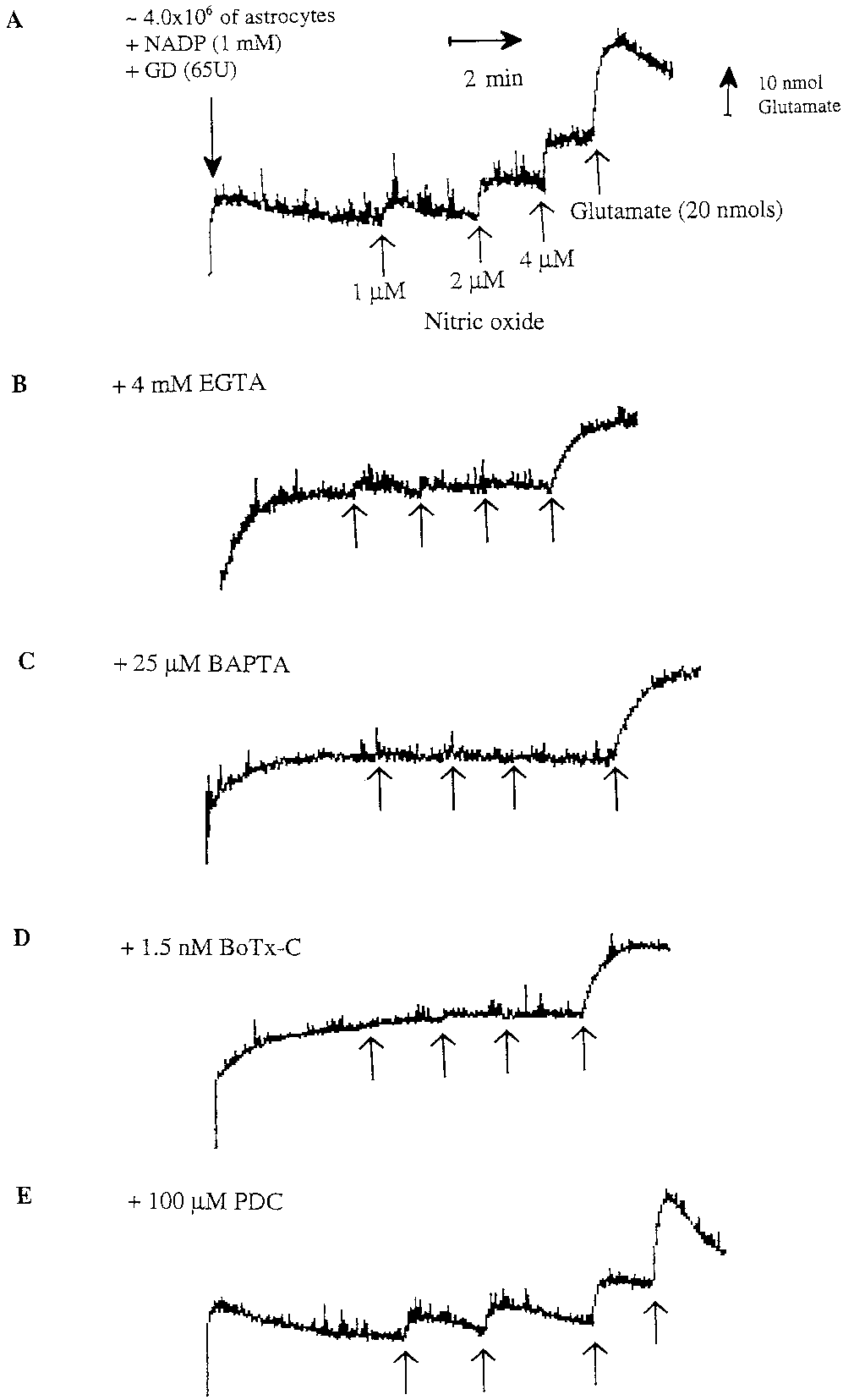 NO Induces Rapid Calcium-Dependent Release
NO Induces Rapid Calcium-Dependent Release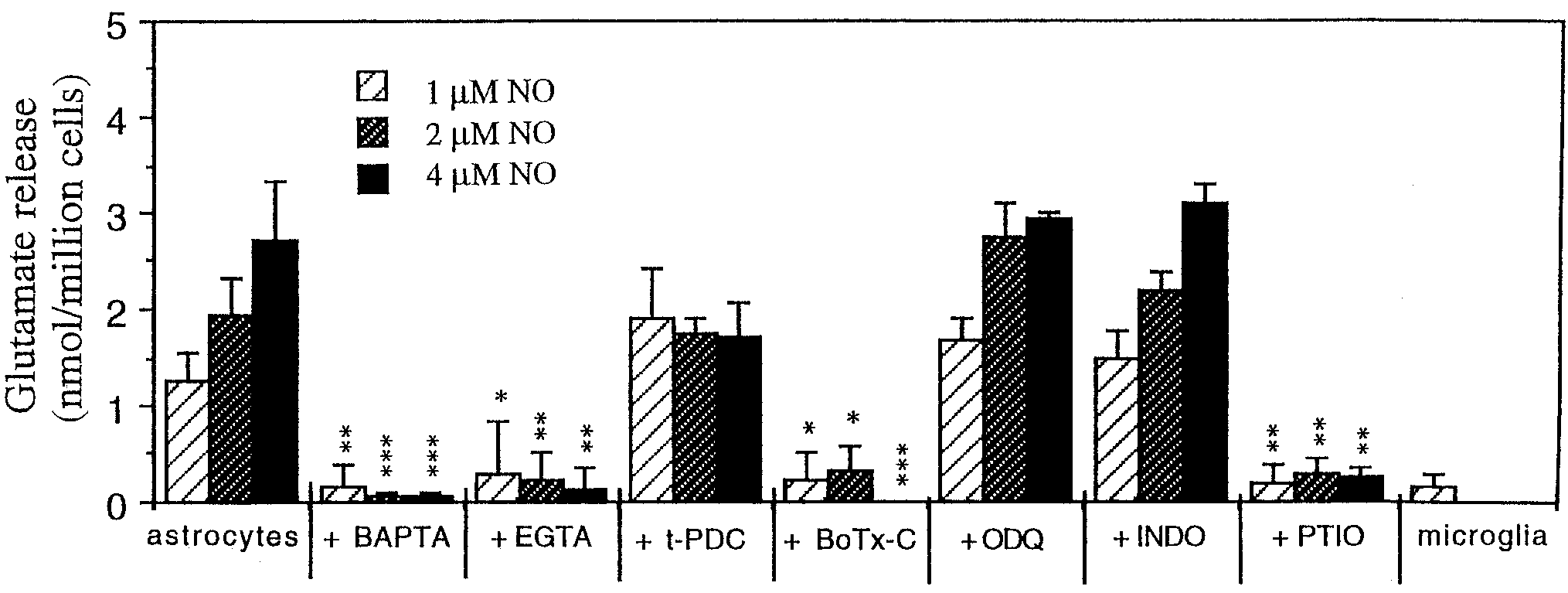
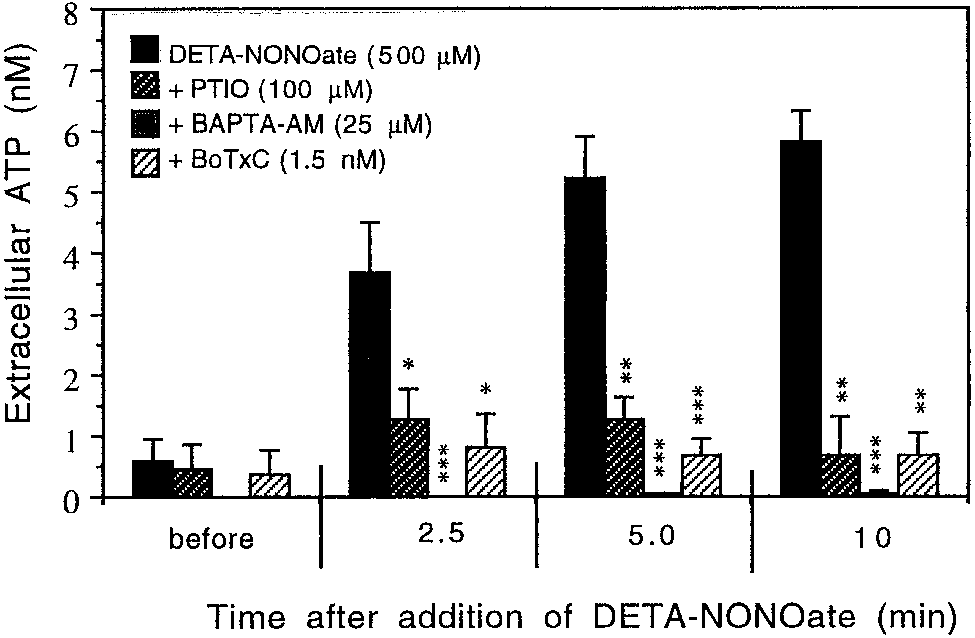 Fig. 3. Quantification of glutamate release evoked by 1, 2, or 4 M
icant glutamate release was seen in response to 1, 2, or 4 M NO.
Fig. 3. Quantification of glutamate release evoked by 1, 2, or 4 M
icant glutamate release was seen in response to 1, 2, or 4 M NO.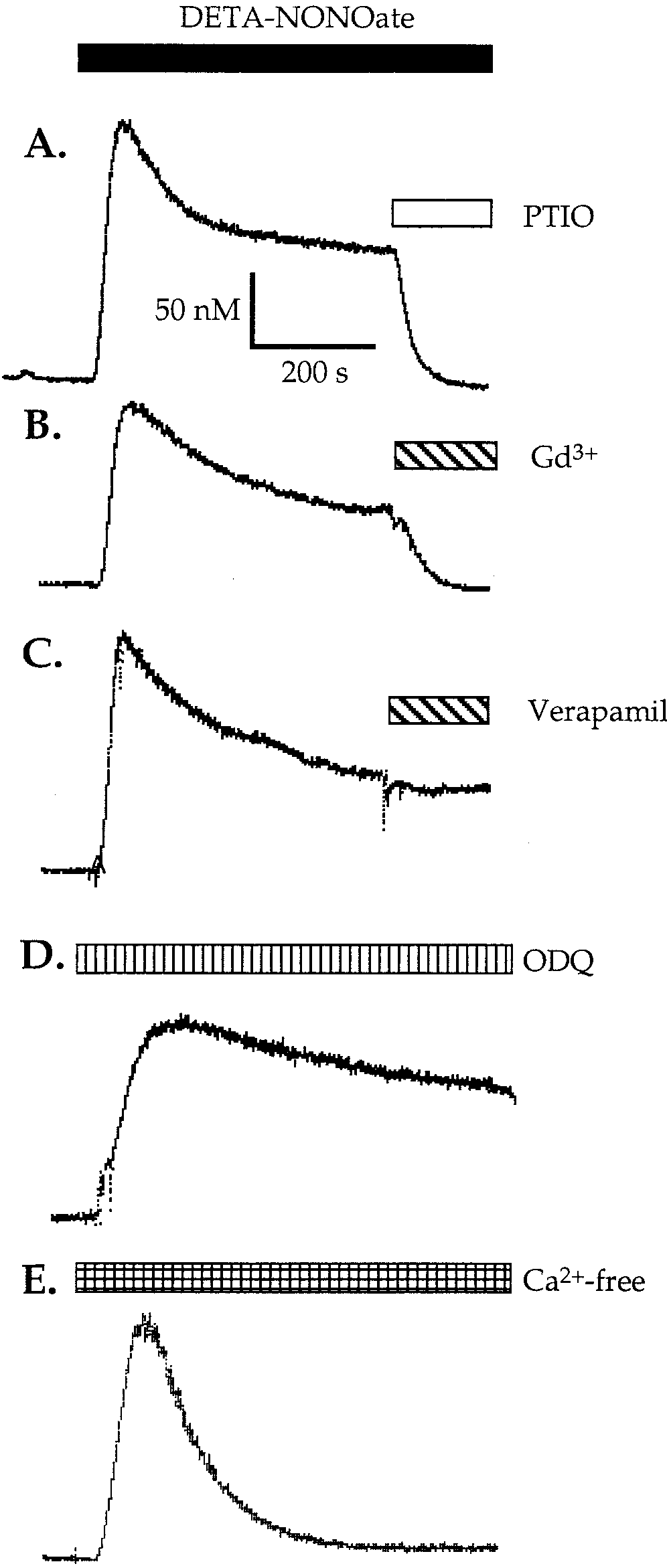 glutamate addition (Fig. 7). After 24 h, glutamate was
a steady-state level lower than in the absence of 1400W
still present in the medium of activated astrocytes
(but higher than in nonactivated astrocytes).
glutamate addition (Fig. 7). After 24 h, glutamate was
a steady-state level lower than in the absence of 1400W
still present in the medium of activated astrocytes
(but higher than in nonactivated astrocytes).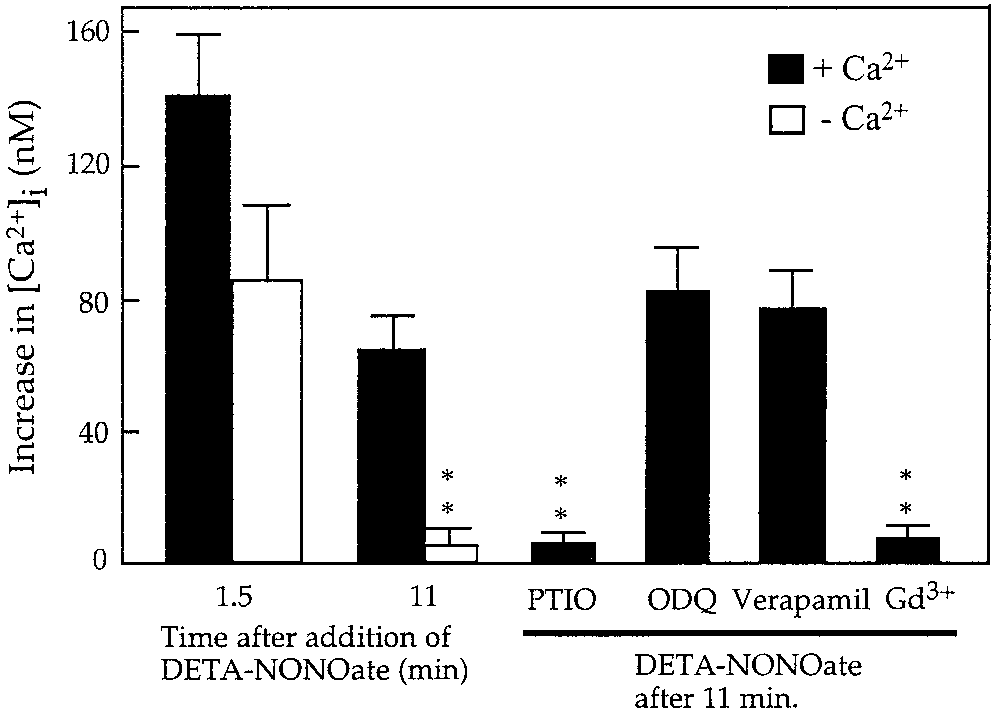
 nisms are not relevant in astrocytes, at least in ourconditions.
nisms are not relevant in astrocytes, at least in ourconditions.